



Анемия у пациентов с хронической сердечной недостаточностью: как подобрать оптимальную терапию?
 Всемирной организацией здравоохранения (ВОЗ) определено, что критерием анемии является концентрация гемоглобина (Нb) в крови <13 г/дл у мужчин и <12 г/дл у женщин.
Всемирной организацией здравоохранения (ВОЗ) определено, что критерием анемии является концентрация гемоглобина (Нb) в крови <13 г/дл у мужчин и <12 г/дл у женщин.
По данным литературы, частота анемии колеблется от 16 до 48%, возрастая по мере увеличения класса по NYHA [1-4].
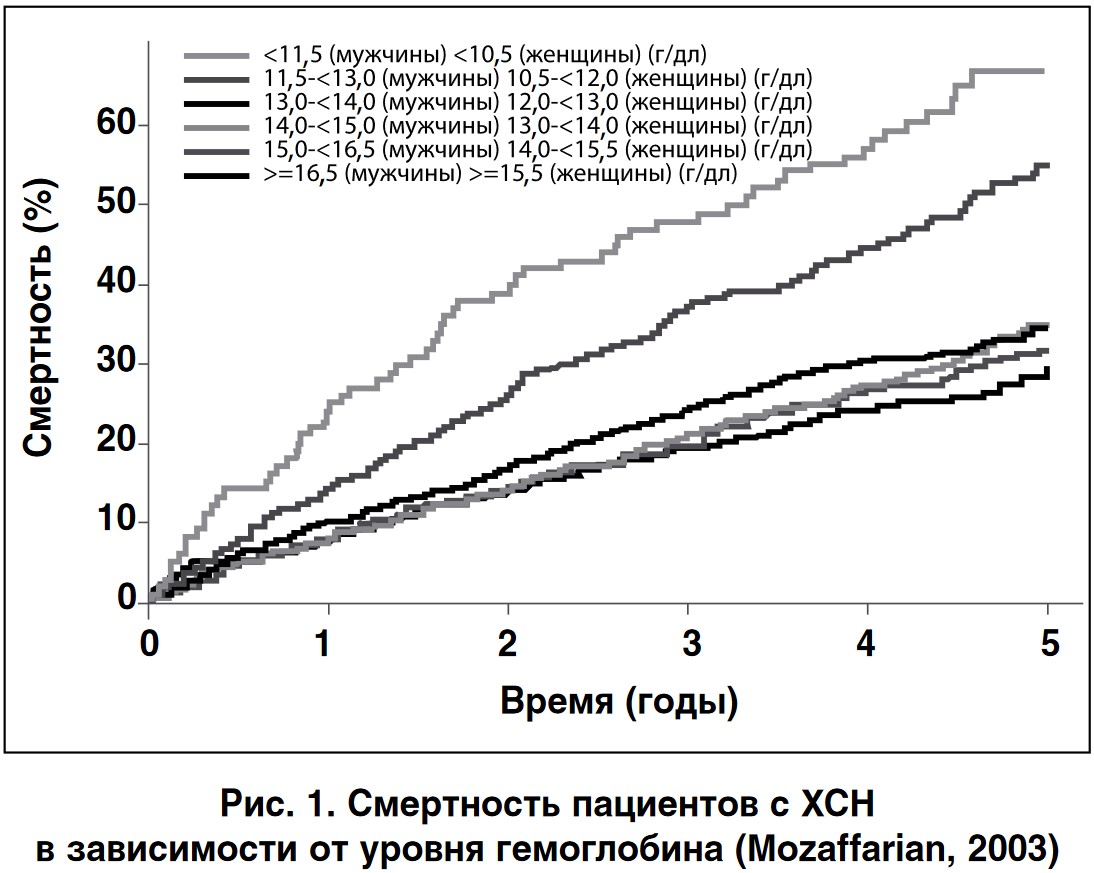
Анемия оказывает существенное негативное влияние на клинический прогноз пациентов с хронической сердечной недостаточностью (ХСН) (рис. 1) а также снижает функциональные возможности и качество жизни пациентов [5, 6].
В целом можно говорить о многофакторном генезе феномена анемии при ХСН, и для лучшего понимания механизмов ее возникновения необходимо кратко рассмотреть ключевые факторы, принимающие участие в регуляции уровня Нb в крови.
Эритропоэтин (ЭП). Представляет собой гликопротеин, 80% которого синтезируется в почках перитубулярными эндотелиоцитами и фибробластами [7, 8], а 20% – гепатоцитами [9]. Главным стимулом к экспрессии и синтезу ЭП является гипоксия почек [2], которая может являться следствием не только снижения ренального кровотока, но и увеличения потребности почек в кислороде, которое сопутствует возрастанию канальцевой реабсорбции Na+ [10]. Существует физиологическая зависимость между степенью снижения уровня Hb и концентрацией циркулирующего ЭП, имеющая экспоненциальный характер [11]. ЭП стимулирует эритропоэз, воздействуя в костном мозге на два типа клеток-предшественников эритроцитов – проэритробласты и CFU-E – через связывание с находящимися на поверхности последних специфическими рецепторами к ЭП [12]. Взаимодействие ЭП с вышеуказанными рецепторами подавляется провоспалительными цитокинами [13].
Железо (Fe) и его метаболизм. Метаболизм ионизированного Fe, требующегося для синтеза Hb в процессе эритропоэза, представляет собой своего рода замкнутый цикл. Из 12-18 г Fe, поступающего за сутки в организм с пищей, всасываются (в duodenum и jejunum) не более 5-10%, что в среднем соответствует 1 мг. Средняя суточная потеря Fe соответствует тому же 1 мг и происходит за счет отмирающих клеток слизистых и кожи, а также микропотерь крови [14]. Общее содержание Fe в крови является стабильным, колеблется у взрослых от 3 до 5 г (40-60 мг/кг массы тела) и распределяется на его функциональный (т. е. активно используемый) пул (80%) и пул депонированного Fe (в составе ферритина и гемосидерина – 20%).
Доминирующий функциональный пул представлен железом: а) используемым для синтеза Hb (примерно 2/3 от всего содержащегося в организме); б) находящимся в составе энзимов, главным образом, дыхательной цепи (примерно 10%); в) находящимся в составе мышечного белка миоглобина (примерно 5%) и г) связанным с белком трансферрином, выполняющим роль переносчика Fe.
Пул депонированного Fe пребывает в связанном состоянии в составе белка ферритина, основным местом нахождения которого являются макрофаги ретикулоэндотелиальной системы – РЭС (селезенка, костный мозг). Меньшее количество связанного Fe находится в печени и клетках слизистой оболочки кишечника [15]. После захвата и последующего транспортирования через энтероциты Fe в виде трехвалентной формы поступает в кровь, где связывается с трансферрином, осуществляющим его транспорт в костный мозг. В костном мозге трансферрин взаимодействует со своими специфическими рецепторами (TfR) на поверхности эритроидных клеток-предшественников, в результате чего достигается превращение Fe3+ в ионы Fe2+ с последующей инкорпорацией последних в цитозоль и проникновением в митохондрии, являющиеся местом синтеза гема. Часть транспортируемого Fe через аналогичный TfR-опосредуемый механизм инкорпорируется в гепатоциты для депонирования в печени. Вместе с тем трансферрин практически не участвует в инкорпорации Fe макрофагами РЭС, поскольку последние обеспечивают эту функцию самостоятельно, поглощая и расщепляя Hb, высвобождающийся в результате естественной гибели достигших старости эритроцитов [16].
Концентрация трансферрина в плазме возрастает соответственно степени истощения пула депонированного Fe в организме и, напротив, снижается при его увеличении. Однако определение данного показателя не имеет достаточной диагностической ценности, поскольку в условиях системного восполнения синтез трансферрина может подавляться [17]. Напротив, высокой диагностической ценностью обладает показатель сатурации (насыщения) трансферрина железом, который отражает долю задействованных активных сайтов связывания Fe3+ на поверхности его молекулы от общего их числа. Сатурация трансферрина <20% является информативным показателем дефицита Fe в организме независимо от его причины.
Ферритин представляет собой белок, находящийся в связанном с трехвалентным Fe состоянии в клетках РЭС и печени, а также в относительно небольших концентрациях – в системном кровотоке. Диагностическую ценность представляет существенное (<30 мг/л) снижение уровня ферритина в плазме, свидетельствующее о так называемом абсолютном железодефиците, а уровни <10-15 мг/л отражают практически полное отсутствие депонированного Fe в организме. Повышенные и даже нормальные уровни ферритина имеют ограниченное диагностическое значение, поскольку последний одновременно является белком острой фазы воспаления, способным реагировать на различные, в том числе системного характера, иммуновоспалительные процессы. Повышенный или нормальный уровень ферритина также может быть следствием нарушения функции печени, в которой происходит его деградация [15].
Существенное значение в регуляции обмена Fe и, соответственно, в патогенезе анемии играет гепсидин. Последний представляет собой синтезируемый в печени пептид, главная роль которого состоит в угнетении высвобождения депонированного Fe3+. Синтез гепсидина уменьшается при снижении концентрации Hb, истощении запасов Fe в организме и, напротив, возрастает при его избыточном поступлении (например, частых гемотрансфузиях). Важным обстоятельством является то, что экспрессия гепсидина существенно возрастает при системных иммуновоспалительных состояниях, к числу которых, в соответствии с современными представлениями, принадлежит и ХСН. Прямыми индукторами экспрессии гепсидина являются провоспалительные цитокины – интерлейкин‑1, интерлейкин‑6, TNF [17, 18].
Возможные механизмы развития анемии у пациента с ХСН
1. Ренальный механизм. Существенное снижение скорости клубочковой фильтрации (СКФ) <60 мл/мин/1,72 м2 наблюдается у 50-57% пациентов с ХСН [19, 20]. Классическая схема анемии почечного генеза предполагает снижение ЭП-синтезирующей функции почек, пропорциональное выраженности повреждения последних, что влечет за собой уменьшение эритропоэза [21]. Вышеуказанный ренальный, связанный со снижением выработки ЭП, механизм становится актуальным при уменьшении СКФ <35-40 мл/мин/1,72 м2 [22].
У пациентов с ХСН снижение ренального кровотока приводит к усилению выработки ЭП через механизм гипоксии [20], в связи с чем у большинства пациентов с ХСН и анемией определяются не сниженные, а, напротив, повышенные либо нормальные уровни ЭП [23, 24, 38]. Данную ситуацию трактуют как резистентность к эндогенному ЭП, в основе которой лежат десенситизация рецепторов к ЭП, обусловленная влиянием воспалительных цитокинов [13], дисфункция таргетных клеток костного мозга на фоне его хронической гипоксии [25], прямое угнетение пролиферации последних воспалительными цитокинами [26]. В то же время для пациентов с ХСН и тяжелой ренальной дисфункцией характерным является снижение ЭП-образующей функции последних [27]. Поэтому термин «кардиоренальный анемический синдром» [28] применим именно к категории пациентов с анемией с сопутствующим тяжелым нарушением азотовыделительной функции почек.
2. Иммуновоспалительный механизм. Последний играет ведущую роль в подавлении эритропоэза в условиях хронических инфекций, онкологических заболеваний и при некоторых других состояниях, характеризующихся системной иммуновоспалительной активацией (ревматоидный артрит, диффузные болезни соединительной ткани, сахарный диабет и др., а также ХСН). В вышеозначенном механизме главным «действующим лицом» выступают провоспалительные цитокины (TNF, интерлейкины 1 и 6, гамма-интерферон, бактериальные липополисахариды), те или иные из которых прямо либо опосредованно: 1) блокируют транспорт Fe внутри энтероцита в кровь; 2) угнетают высвобождение депонированного Fe из РЭС (ретикулоэндотелиальный блок) и гепатоцитов [18]. Ключевым посредником такого рода блокады выступает синтезируемый в печени гепсидин, секреция которого стимулируется провоспалительными цитокинами [30], а также застойными явлениями в печени [31]. В результате возникает так называемый функциональный дефицит Fe, состоящий в том, что при наличии его достаточных запасов наблюдается его нехватка для эритропоэза [17]. Наконец, провоспалительные цитокины играют ведущую роль в формировании резистентности к эндогенному ЭП (см. выше).
Анемия, наблюдающаяся на фоне функционального железодефицита в условиях системного воспаления, терминологически определена как анемия хронического заболевания (АХЗ). Для данного состояния, наряду с уменьшением содержания Fe в крови и сниженной сатурацией железом трансферрина, характерен нормальный или повышенный уровень ферритина. В пользу АХЗ могут также свидетельствовать повышенные уровни циркулирующих биомаркеров воспаления (С-реактивный протеин, воспалительные цитокины) [15].
Для АХЗ более характерен нормоцитоз – средний корпускулярный объем эритроцита (MCV) находится в пределах 80-100 фл, однако у 1/5-1/3 таких пациентов анемия носит характер микроцитарной (показатель MCV <80 фл). Доля АХЗ в структуре всех случаев анемии при ХСН достигает 60% [32, 37].
3. Истинный (абсолютный) железодефицит. При ХСН он может быть связан с нарушением кишечной абсорбции Fe и микропотерями крови. К возможным причинам нарушения всасывания Fe у пациентов с ХСН относят отек и утолщение стенок кишечника, снижение кровотока в органах брюшной полости [33], анорексию и недостаточное питание [34]. Микропотери крови, роль которых у пациентов с ХСН, возможно, недооценивается, могут быть связаны с регулярным приемом большинством из них ацетилсалициловой кислоты либо антикоагулянтов [35, 36].
Частота истинного железодефицита среди пациентов с ХСН и сопутствующей анемией колеблется, по разным данным, от 5 до 21% [20, 32, 37, 38]. Для абсолютного железодефицита, помимо сниженной сатурации трансферрина и повышения содержания в плазме его растворимых рецепторов, характерным является снижение концентрации циркулирующего ферритина. Хотя традиционным критерием абсолютного железодефицита считается уровень ферритина <30 мкг/л, для пациентов с ХСН в последнее время в качестве такого критерия рассматривается величина <100 мкг/л [39, 40]. Это обусловлено результатами оценки уровней ферритина у пациентов с тяжелой ХСН в зависимости от наличия у них истощения депо Fe по данным пункционной биопсии костного мозга. Так, у пациентов с истощением запасов Fe средний уровень ферритина в крови составил 75 мкг/л, а у пациентов с сохранным пулом депонированного Fe – 211 мкг/л [41].
Истинная железодефицитная анемия является микроцитарной (показатель MCV <80 фл) и гипохромной (среднее содержание Hb в эритроците – МСН <27 пг).
4. Гемодилюция. Увеличенный объем циркулирующей плазмы, характерный для гиперволемии, наблюдающейся в фазе декомпенсации ХСН, снижает гематокрит и, соответственно, показатель концентрации Hb в крови – так называемая псевдоанемия, о которой представляется возможным говорить лишь в случаях, когда достижению эуволемии сопутствует нормализация уровня Hb без применения специфической терапии. Вместе с тем вышеозначенный гемодилюционный механизм, по-видимому, может принимать участие в формировании показателя Hb крови и у многих клинически стабильных пациентов с ХСН. Об этом могут свидетельствовать результаты исследований, в которых с помощью современных радиоизотопных методов было продемонстрировано увеличение объема циркулирующей плазмы у 40-50% пациентов с ХСН, не имевших на момент исследования явных объективных признаков гиперволемии [43, 44].
5. Дефицит витамина В12 или фолиевой кислоты. Удельный вес макроцитарной анемии, связанной с вышеуказанным дефицитом, относительно невысок (5-8%) [32, 37] и, как правило, сопряжен с соответствующими коморбидными состояниями (алкоголизм, парентеральное питание, заболевание либо резекция желудка или тонкого кишечника и т. п.).
6. Влияние лекарственных препаратов. Помимо препаратов, способных обусловить повреждение слизистых желудочно-кишечного тракта (ЖКТ) и тем самым спровоцировать микро- или макрокровопотерю (ацетилсалициловая кислота, нестероидные противовоспалительные средства), следует упомянуть ряд лекарств, применению которых сопутствует риск подавления эритропоэза – цитостатики, некоторые антибактериальные средства. Наибольшее внимание привлекают данные о способности ингибиторов ангиотензинпревращающего фермента (ИАПФ) подавлять эритропоэз [45, 46]. Это, в частности, объясняют тем, что АПФ принимает активное участие в биодеградации белка Ас-SDKP – мощного ингибитора гемопоэза [23]. Вместе с тем на сегодняшний день клинические исследования, убедительно подтверждающие связь приема ИАПФ со снижением уровня Hb, отсутствуют, а наличие анемии в действующих рекомендациях не относится к числу противопоказаний к назначению данного класса препаратов.
Вышеизложенное наглядно демонстрирует многофакторный характер анемии при ХСН. Это естественным образом предполагает возможность сочетания различных механизмов ее формирования у одного и того же пациента. Вместе с тем уточнение вышеозначенных механизмов анемии в каждом конкретном случае остается актуальным. Оправданным выглядит использование следующего практического алгоритма уточнения природы анемии (рис. 2).
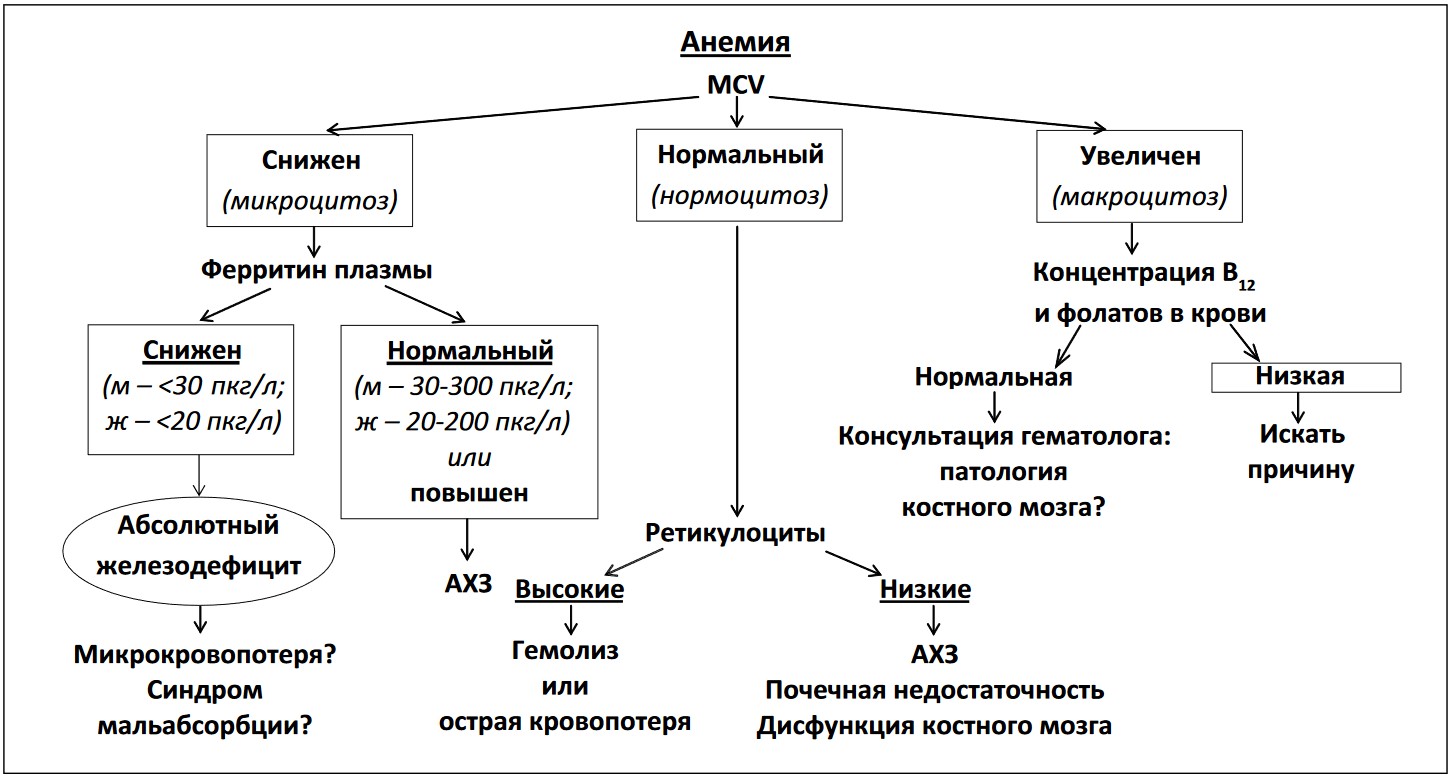
Рис. 2. Схема диагностического поиска при анемии (модифицировано по Anker, von Hoehling) [15]
Примечания.
Концентрация железа в сыворотке крови очень изменчива с высокой вариабельностью в течение суток; этот показатель не должен использоваться для диагностики дефицита железа [46]. MCV – средний корпускулярный объем эритроцита; АХЗ – анемия хронического заболевания; м – мужчины; ж – женщины.
Учитывая потенциально сложную природу анемии при ХСН, у данной категории пациентов понятия «ренальная анемия», «АХЗ», «дилюционная анемия» вряд ли должны звучать как диагностические термины. Однако точная оценка природы анемии, безусловно, представляется полезной для правильного подхода к ее коррекции.
До недавнего времени в качестве оправданной рассматривалась стратегия лечения анемии при ХСН с помощью эритропоэзостимулирующих агентов, в том числе используемых в комбинации с препаратами Fe. В ряде рандомизированных клинических испытаний, выполненных начиная с 2000 г., было установлено, что применение рекомбинантного ЭП (эпоэтин) у пациентов с ХСН и анемией способно восстанавливать уровень Hb до целевых значений с сопутствующими благоприятными клиническими эффектами в виде снижения класса по NYHA, увеличения толерантности к физической нагрузке, улучшения параметров качества жизни, улучшения систолической функции левого желудочка [48-51]. Клинические испытания дарбэпоэтина-α (эритропоэзостимулирующий протеин, взаимодействующий с теми же, что и ЭП, рецепторами, однако обладающий более выгодными в сравнении с последним фармакологическими характеристиками) продемонстрировали сходные результаты [52-54].
Вместе с тем еще с конца 1990-х гг. начали накапливаться данные о повышении риска сердечно-сосудистых осложнений, в том числе фатальных, на фоне использования эритропоэзостимулирующих препаратов. Так, крупное исследование NHCT (1998), включившее более 1 тыс. гемодиализных пациентов с сопутствующей ишемической болезнью сердца или ХСН, было прекращено досрочно из-за соображений безопасности в связи с выявленным статистически достоверным трендом в сторону увеличения частоты случаев смертности [55]. Лечение эпоэтином-α пациентов с хронической болезнью почек (ХБП) сопровождалось в исследовании CHOIR достоверным, на 34%, повышением частоты достижения комбинированной конечной точки (смерть, госпитализация по поводу СН, инфаркт миокарда или инсульт) в сравнении с плацебо [56]. Наконец, в исследовании TREAT, охватившем более 4 тыс. пациентов с ХБП и анемией, треть из которых имели СН, применение дарбэпоэтина ассоциировалось с почти двукратным риском развития мозгового инсульта [57].
Конец эры эритропоэтиностимулирующих средств при ХСН наступил после окончания широкомасштабного (2278 пациентов) исследования RED-HF, в котором использование дарбэпоэтина у пациентов с систолической ХСН и уровнем Hb от 9,0 до 12,0 г/дл, хотя и сопровождалось, в сравнении с плацебо, нормализацией уровня Hb и улучшением качества жизни, было ассоциировано с достоверным (р=0,01) увеличением частоты тромбоэмболических осложнений [58]. Более того, в выполненном в США и обнародованном в 2014 г. ретроспективном обсервационном исследовании, включившем более 2 тыс. пациентов с ХСН и ренальной дисфункцией (СКФ <60 мл/мин/1,73 м2), лечение ЭП ассоциировалось с увеличением риска смерти на 40% (р=0,02), почти двукратным высокодостоверным (р<0,001) возрастанием риска возникновения острого коронарного синдрома наряду с достоверным увеличением количества госпитализаций [59].
В качестве возможных причин вышеуказанных неутешительных результатов рассматривается повышение гематокрита (и, соответственно, вязкости крови), влекущее за собой возрастание риска тромбоза, а также повышение артериального давления у пациентов с артериальной гипертензией, нередко сопутствующее применению ЭП [60, 61]. Так или иначе, в сложившейся ситуации для лечения хронической немакроцитарной анемии при ХСН у клиницистов осталось единственное «оружие» в виде препаратов Fe. Последние представлены пероральными и внутривенными (в/в) формами.
Пероральные формы
Их преимуществом является относительно невысокая стоимость, а главным недостатком – низкая всасываемость. До последнего времени пероральные препараты Fe были представлены исключительно двухвалентными формами в виде сульфатной и глюконатной солей. Их существенный недостаток состоит в том, что окислению Fe2+ в Fe3+ в слизистой кишечника сопутствует образование свободных радикалов, оказывающих локальное токсическое действие. Оксидантный стресс клеток ЖКТ является причиной весьма частых, наблюдающихся примерно у 60% пациентов, гастроинтестинальных жалоб в виде изжоги, тошноты, запоров или диареи, которые сопровождают применение пероральных препаратов двухвалентного Fe [62]. Переносимость последних улучшается при совмещении их приема с приемом пищи, однако такой подход, в свою очередь, существенно ухудшает их всасываемость, что обусловливает значительные трудности в достижении баланса между минимизацией побочных эффектов и максимализацией усвояемости препарата Fe2+ [63]. Значение оксидантного стресса ЖКТ, провоцируемого поступлением экзогенного Fe2+, не ограничивается желудочно-кишечным дискомфортом как таковым. Согласно данным экспериментальных исследований длительный прием солей двухвалентного Fe через механизм оксидантного стресса может вызывать деструкцию клеточных мембран, повреждение ДНК и иммунные нарушения [64].
Через тот же механизм вышеуказанные соли усугубляют выраженность экспериментального колита, негативно влияют на кишечную микрофлору, а также могут увеличивать риск развития железистых опухолей кишечника [65, 66].
Вышеуказанных последствий, связанных со свободно-радикальным повреждением ЖКТ, возможно избежать при применении трехвалентного железа в виде перорального Fe3+ – полимальтозного комплекса; он содержит полимальтозную оболочку, обеспечивающую контролируемое высвобождение Fe и одновременно минимизирующую его контакт с пищей и другими лекарствами [67, 68].
Универсальным ограничением пероральной ферротерапии является ее малодейственность при патологических состояниях, сопровождающихся нарушениями всасывания Fe, которые имеют место у большинства больных с клинически выраженной ХСН.
В качестве причин недостаточной абсорбции Fe у таких пациентов рассматриваются отек слизистой кишечника [69] и сниженный (на 30-43%) кишечный кровоток [70]. Кроме того, при АХЗ, механизм которой актуален для ХСН, повышенные уровни гепсидина угнетают транспортировку ферропортином железа внутри энтероцита, целью которой является поступление Fe в кровь [71]. К дополнительным факторам, способным ограничить всасывание препаратов Fe в кишечнике, относятся прием некоторых медикаментов (например, блокаторов Н2-рецепторов), паретические расстройства со стороны желудка, нередко наблюдающиеся при диабете [72].
Наиболее существенным недостатком стратегии коррекции железодефицита с помощью пероральных форм Fe является недостаточная скорость восполнения ими запасов Fe. Так, суточная доза Fe, требующаяся для восполнения его дефицита, составляет 1000 мг.
Биодоступность пероральных форм Fe составляет примерно 10% [73]. Исходя из обычной дозы сульфата железа (100-200 мг/сут), в том «идеальном» случае, если пациент адекватно переносит дозу в 200 мг и не имеет предпосылок к нарушению всасывания Fe, ежедневное восполнение его запаса составит 20 мг, что потребует минимум 50 дней для устранения железодефицита. При менее благоприятном сценарии – если это пациент с выраженной ХСН (при которой абсорбция Fe снижена примерно на 50%), который способен переносить лишь дозу 100 мг/сут – ежедневное восполнение Fe составит не более 5 мг, что потребует 200 дней для полного восстановления его запасов. В реальной практике для этого может потребоваться еще большее время из-за спорадических пропусков очередной дозы по случайным причинам или из-за желудочно-кишечного дискомфорта [63]. Приведенные расчеты свидетельствуют, что надлежащая результативность пероральной ферротерапии может быть достигнута при условии ее длительности более 6 мес. В то же время для полного восстановления запасов Fe достаточно от одной до нескольких его в/в инъекций. Поэтому в/в ферротерапия сегодня рассматривается в качестве приоритетного подхода к коррекции железодефицита и сопряженной с ним анемии.
Внутривенные препараты железа
Современные в/в препараты Fe воплощены в виде сфероподобных коллоидных микрочастиц железо-углеводной природы. Ядро такой микрочастицы содержит Fe3+ в виде оксигидроксидного комплекса, его окружает углеводная оболочка различной (в зависимости от запатентованной лекарственной формы) природы, обеспечивающая микрочастице стабильность в растворе и в кровотоке [74]. Введенные в/в вышеуказанные частицы инкорпорируются фагоцитами РЭС, которые разрушают их оболочку и далее выбрасывают ионы трехвалентного Fe в кровоток, где они немедленно связываются с трансферрином для последующего транспортирования в костный мозг.
Экспорт Fe из микрофагов в кровоток осуществляется транспортным белком ферропортином. При функциональном дефиците Fe, свойственном иммуновоспалительному механизму анемии (при АХЗ), повышенные уровни гепсидина вызывают деградацию ферропортина и, соответственно, задержку Fe внутри макрофагов (гепсидиновый, или ретикулоэндотелиальный, блок). После в/в инъекции препаратов Fe наблюдается быстрое нарастание его концентрации внутри макрофагов, что индуцирует гиперэкспрессию ферропортина, и тем самым гепсидиновый блок преодолевается [75]. Именно этим механизмом объясняется эффективность в/в форм Fe при его относительном (функциональном) дефиците.
В настоящее время на европейском рынке доступны четыре в/в лекарственные формы Fe, различающиеся природой углеводной оболочки, дозированием и технологией назначения, – железа декстран, глюконат железа, сахарат железа и карбоксимальтозат железа.
Первые две формы, особенно железа декстран, характеризуются меньшей безопасностью применения в сравнении с последними двумя формами. Так, согласно базе данных Управления по контролю качества продуктов питания и лекарственных средств США (FDA), охватившей период 1997-2002 гг., при применении декстрана железа наблюдали 29,2 случая угрожающих жизни анафилактических реакций на 1 млн пациентов, глюконата железа – 10,5, сахарата железа – 4,2 [76]. В ныне действующих в США рекомендациях по лечению пациентов с ХБП говорится о нежелательности использования в/в декстрана железа [77]. На сегодняшний день база данных, касающаяся эффективности в/в терапии препаратами Fe пациентов с ХСН, представлена результатами четырех рандомизированных испытаний сахарата железа и карбоксимальтозата железа, выполненных в 2007-2014 гг.
В первом из них участвовали 40 пациентов (20 – активный препарат, 20 – плацебо) с фракцией выброса <35% и анемией (Hb <12,5 г/дл – у мужчин и <11,5 г/дл – у женщин) при концентрации у них ферритина в плазме <100 пк/мл и насыщении трансферрина железом <20%.
Лечение заключалось во в/в введении 200 мг железа (сахарат железа) 1 раз в неделю в течение 5 нед, срок наблюдения пациентов составил 26 нед. К концу наблюдения в группе активного препарата в отличие от плацебо наблюдали достоверное (р<0,01) улучшение симптоматики и параметров качества жизни, снижение класса по NYHA, повышение переносимости нагрузки, снижение концентрации циркулирующего NT-pro-BNP, чему сопутствовала нормализация уровня Hb, содержания ферритина и насыщения трансферрина [80].
В следующее, обнародованное в 2008 г., испытание того же сахарата железа было включено 35 пациентов с ХСН с различным уровнем Hb и дефицитом Fe, в качестве критериев которого были установлены уровень ферритина <100 нг/мл (абсолютный железодефицит) или 100-300 нг/мл при насыщении трансферрина <20% (относительный железодефицит) – так называемые прагматические критерии FERRIC-HF, ставшие ориентиром для последующих клинических протоколов. Схема терапии состояла во введении 200 мг препарата еженедельно до достижения уровня ферритина 500 нг/мл и затем 200 мг ежемесячно (поддерживающий этап). В конце 18-месячного периода наблюдения в группе активного лечения наблюдали достоверное возрастание уровня ферритина и сатурации трансферрина, увеличение максимального потребления кислорода на пике нагрузки и времени ее выполнения наряду с достоверным снижением класса по NYHA [81].
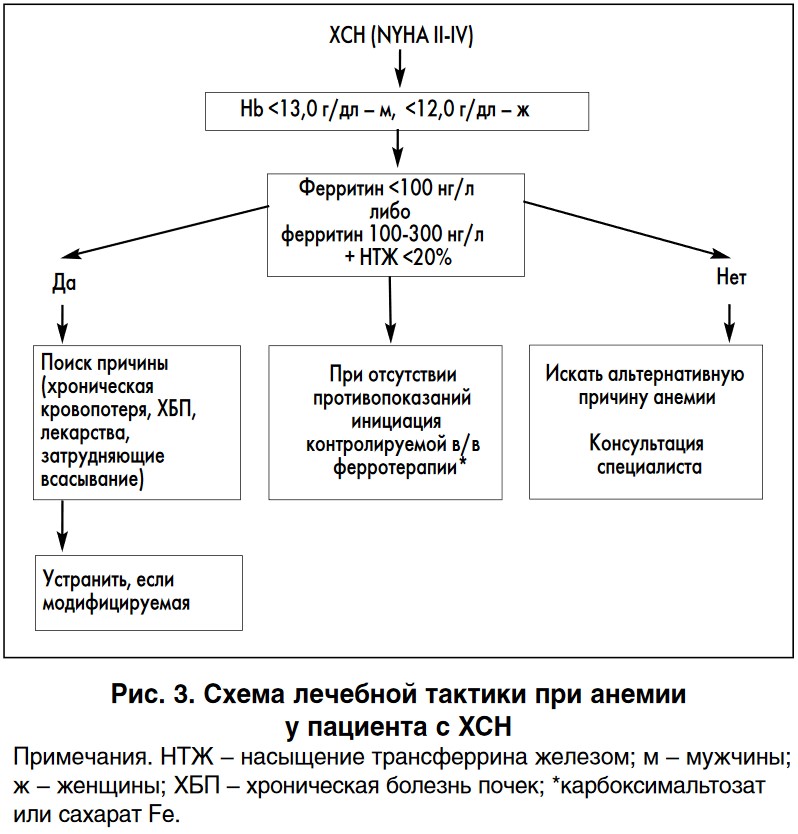 В свете вышеизложенного может быть рекомендован представленный на рисунке 3 алгоритм коррекции анемии, сопряженной с абсолютным или функциональным железодефицитом у пациентов с ХСН.
В свете вышеизложенного может быть рекомендован представленный на рисунке 3 алгоритм коррекции анемии, сопряженной с абсолютным или функциональным железодефицитом у пациентов с ХСН.
В заключение следует еще раз подчеркнуть, что для анемии при ХСН характерно многообразие механизмов, которые, по всей видимости, могут параллельно участвовать в ее формировании.
Концепция АХЗ как едва ли не самого актуального механизма анемии при ХСН хорошо обоснована и выглядит привлекательно, однако оставляет открытым вопрос о том, почему у значительной части пациентов с клинически выраженной, прогрессирующей ХСН уровень Hb остается нормальным вплоть до финального этапа заболевания. Предстоящие исследования должны дать ответ на этот вопрос.
Наличие при ХСН хронической анемии, сопряженной с лабораторными признаками абсолютного либо относительного железодефицита, должно рассматриваться в качестве сигнала к инициации контролируемой в/в ферротерапии, предпочтительно сахаратом или карбоксимальтозатом железа. Важность такой терапии определяется предоставляемыми ею возможностями существенного и стойкого улучшения самочувствия, качества жизни и клинико-функционального состояния таких пациентов.
Литература
1. Horwich T.B., Fonarow G.C., Hamilton M.A., et al. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure // J. Am. Coll. Cardiol. – 2002. – Vol. 39. – Р. 1780-1786.
2. Kosiborod M., Smith G.L., Radford M.J., et al. The prognostic importance of anemia in patients with heart failure // Am. J. Med. – 2003. – Vol. 114. – Р. 112-119.
3. Anand I.S., Kuskowski M.A., Rector T.S., et al. Anemia and change in hemoglobin over time relatеs to mortality and morbidity in patients with chronic heart failure: results from ValHeFT // Circulation. – 2005b. – Vol. 112. – Р. 1121-1127.
4. Komajda M., Anker S.D., Charlesworth A., et al. The impact of new onset anemia on morbidity and mortality in chronic heart failure: results from COMET // Eur. Heart J. – 2006. – Vol. 27. – Р. 1440-1446.
5. Kalra P.R., Bolger A.P., Francis D.P., et al. Effect of anemia on exercise tolerance in chronic heart failure in men // Am. J. Cardiol. – 2003. – Vol. 91. – Р. 888-891.
6. Воронков Л.Г., Паращенюк Л.П., Яновський Г.В. та ін. Предиктори якості життя у хворих з хронічною серцевою недостатністю ІІІ функціонального классу за NYHA // Серце і судини. – 2009. – № 1. – С. 81-85.
7. Koury S.T., Bondurant M.C., Koury M.J. Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization // Blood. – 1988. – Vol. 71. – Р. 524-537.
8. Donnley S. Why is erythropoietion made in the kidney? The kidney functions as a critmeter // Am. J. Kidney. Dis. – 2001. – Vol. 38. – Р. 415-425.
9. Koury S.T., Bondurant M.C., Koury M.J., et al. Localisation of cells producing erythropoietin in murine liver in situ hybridization // Blood. – 1991. – Vol. 77. – Р. 2497-2503.
10. Jelkman W. Molecular biology of erythropoietin // Int. Med. – 2004. – Vol. 43. – Р. 649-659.
11. Thomas M.C. Anemia in diabetes: marker or mediator of microvascular disease? // Nat. Clin. Pract. Nephrol. – 2007. – Vol. 3. – Р. 20-30.
12. Fisher J.W. Erythropoietin: physiology and pharmacology update // Exp. Biol. Med. – 2003. – Vol. 228. – Р. 1-14.
13. Smrzova J., Balla J., Barany P. Inflammation and resistance to erythropoiesis-stimulating agents – what do we know and what needs to be clarified? // Nephrol. Dial. Translant. – 2005. – Vol. 20 (Suppl 8). – Р. 2-7.
14. Andrews N.C. Disorders of iron metabolism // N. Engl. J. Med. – 199. – Vol. 341. – Р. 1986-1995.
15. Anker S.D., von Haehling S. Anemia in chronic heart failure // Bremen, UNI-MED, 2008. – 79 p.
16. Aisen P., Wessling-Resnick M., Leibold E.A. Iron metabolism // Curr. Opin. Chem. Biol. – 1999. – Vol. 3. – Р. 200-206.
17. Weiss G., Goodnough L.T. Anemia of chronic disease // New. Engl. J. Med. – 2005. – Vol. 352. – Р. 1011-1023.
18. Nemeth E., Tuttle M.S., Powelson J., et al. Hepcidin regulates cellular iron efflux by binding to ferroportion and inducing its internalization // J. Science. – 2004. – Vol. 306. – Р. 2051-2053.
19. Amsalem Y., Garty M., Schwartz R., et al. Prevalence of unrecognized renal insufficiency in patients with heart failure // Eur. Heart J. – 2008. – Vol. 29. – Р. 1029-1036.
20. Anand I.S. Heart failure and anemia: mechanisms and pathophysiology // Heart Fail. Rev. – 2008. – Vol. 13. – Р. 377-378.
21. Tong E.M., Nissenson A.R. Erythropoetin and anemia // Semin. Nephrol. – 2001. – Vol. 21. – Р. 190-203.
22. Thomas S., Rampersad M. Anemia in diabetes // Acta Diabetol. – 2004. – Vol. 41 (Suppl. 1). – S13-S17.
23. Meer P., Lok D.J., Januzzi J.L., et al. Adequacy of endogenous erythropoietin levels and mortality in anaemic heart failure patients // Eur. Heart J. – 2008. – Vol. 29. – Р. 1510-1515.
24. George J., Patal S., Wexier D., et al. Circulating erythropoietin levels and prognosis in patients with congestive heart failure: comparison with neurohormonal and inflammatory markers // Arch. Intern. Med. – 2005. – Vol. 165. – Р. 1304-1309.
25. Andrew J.S. The pathophysiological basis of anemia in chronic heart failure // Europ. J. Heart Failure Suppl. – 2003. – Vol. 2/2. – Р. 213-216.
26. Weiss G. Pathogenesis and treatment of anemia of chronic disease // Blood. Rev. – 2002. – Vol. 16. – Р. 87-96.
27. Silverberg D.S., Wexler D., Blum M., et al. The effect of correction of anemia in diabetics and non-diabetics with resistant congestive heart failure and chronic renal failure by subcutaneous erythropoietin and intravenous iron // Nephrol. Dial. Transplant. – 2003. – Vol. 18. – Р. 141-146.
28. Tobbli I.E., Silverberg D.S., Di Gennaro F., еt al. CRAS. Cardio Renal Anemia Syndrome: Basic and Clinical Aspects / Buenos Aires, 2008. – 115 p.
29. Scrutinio D., Passantino A., Santoro D., еt al. The cardiorenal anemia syndrome in systolic heart failure: prevalence, clinical correlates, and long-term survival // Europ. J. Heart Failure. – 2011. – Vol. 13. – Р. 61-67.
30. Nemeth E., Valore E.V., Terrino M., et al. Hepcidin, a putative mediator of anemia of inflammation, is a type-II acute phase protein // Blood. – 2003. – Vol. 101. – Р. 2461-2463.
31. Suzuki T., Hanawa H., Jiamo S., et al. Inappropriate expression of hepcidin by liver congestion contributes to anemia and relative iron deficiency // J. Cardiac. Failure. – 2014. – Vol. 20. – Р. 268-277.
32. Ezekowitz J.A., McAlister F.A., Armstrong P.W. Anemia is common in heart failure and associated with poor outcomes: insights from a cohort of 12 065 patients with new-onset heart failure // Circulation. – 2003. – Vol. 107. – Р. 223-225.
33. Sandek A., Rauchhaus M., Anker S.D., et al. The emerging role of the gut in chronic heart failure // Curr. Opin. Clin. Nutr. Metab. Care. – 2008. – Vol. 11. – Р. 632-639.
34. Арутюнов Г.П. Анемия у больных с ХСН // Сердечная недостаточность. – Том 4. – № 5. – С. 224-228.
35. Silverberg D.S., Wexler D., Jaina A. The importance of anemia and its correction in the management of severe congestive heart failure // Europ. J. Heart Failure. – 2002. – Vol. 4. – Р. 681-686.
36. Parsi A., Kleber F.X. Anemia in heart failure: its diagnosis and management // Europ. J. Heart Failure. – 2003. – Vol. 5. – Р. 3-4.
37. Opasich C., Cazzola M., Scelsi L., et al. Blunted erythropoietin production and defective iron supply for erythropoiesis as major causes of anemia in patients with chronic heart failure // Europ. Heart J. – 2005. – Vol. 26. – Р. 2232-2237.
38. Westernbrink B.D., Voors A.A., de Boer R.A., et al. Bone marrow dysfunction in chronic heart failure patients // Europ. J. Heart Failure. – 2010. – Vol. 12. – Р. 676-684.
39. Jankowska E.A., von Haehling S., Anker S.D., et al. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives // Europ. J. Heart Failure. – 2013. – Vol. 34. – Р. 816-826.
40. Амосова К.М., Царалунга В.М. Залізодефіцит як нова терапевтична мета у хворих із хронічною серцевою недостатністю // Серце і судини. – 2013. – № 4. – С. 19-26.
41. Nanas J., Matsouka C., Karageorgopoulos D., et al. Etiology of anemia in patients with advanced heart failure // J. Am. Coll. Cardiol. – 2006. – Vol. 48. – Р. 2485-2489.
42. Westernbrink B.D., Visser F.W., Voors A.A., et al. Anemia in chronic heart failure is not only related to impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well // Europ. Heart J. – 2007. – Vol. 28. – Р. 166-171.
43. Androne A.-S., Katz S.D., Lund L., et al. Hemodilution is common in patients with advanced heart failure // Circulation. – 2003. – Vol. 107. – Р. 226-229.
44. Adlbrecht C., Kommata S., Hulsmann M., et al. Chronic heart failure leads to an expanded plasma volume and pseudoanemia, but does not lead to a reduction in the body’s red cell volume // Europ. Heart J. – 2008. – Vol. 29. – Р. 2343-2350.
45. Macdougall I.C. The role of ACE inhibitors and angiotensin II receptor in the response to erythropoietin // Nephrol. Dial. Transplant. – 1999. – Vol. 14. – Р. 1836-1841.
46. UNICEF «National Iron Plus Initiative Guidelines for Control of IDA» (2013).
47. Mc Murray J.J.V., Adamopoulos S., Anker S.D., et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure // Europ. Heart J. – 2012.
48. Silverberg D.S., Wexler D., Blum M., et al. The use of subcutaneous erythropoietin and intravenous iron for the treatment of the anemia of severe, resistant congestive heart failure improves cardiac and renal function and functional cardiac class, and markedly reduces hospitalizations // J. Am. Coll. Cardiol. – 2000. – Vol. 35. – Р. 1737-1744.
49. Mancini D.M., Katz S.D., Lang C.C., et al. Effect erythropoietin on exercise capacity in patients with moderate to severe chronic heart failure // Circulation. – 2003. – Vol. 107. – Р. 294-299.
50. Usmanow R.I., Zueva E.B., Silverberg D.S., et al. Intravenous iron erythropoietin for the treatment of iron deficiency anemia in patients with moderate to severe congestive heart failure and chronic kidney insufficiency // J. Nephrol. – 2008. – Vol. 21. – Р. 236-242.
51. Bergmann M.W., Haufe S., von Knobelsdorff-Brenkenhoff F., et al. A pilot study of chronic, low-dose epoetin-β following percutaneous coronary erythropoietin suggests safety, feasibility, and efficacy in patients with symptomatic ischaemic heart failure // Europ. J. Heart Failure. – 2011. – Vol. 13. – Р. 560-568.
52. Ponikowski P., Anker S.D., Szachiewicz J., et al. Effect of darbepoetin alfa on exercise tolerance in anemic patients with symptomatic chronic heart failure: a randomized, double-blind, placebo-controlled trial // J. Am. Coll. Cardiol. – 2007. – Vol. 49. – Р. 753-762.
53. Gihissis J.T., Kourea K., Panou F., et al. Effect of darbepoetin alfa on right and left ventricular systolic and diastolic function in anemic patients with chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy // Amer. Heart J. – 2008. – Vol. 155. – Р. 751-757.
54. Ghali J.K., Abraham W.T., Fonarow G.C., et al. Randomized double-blind trial darbepoetin alfa patients with symptomatic heart failure and anemia // Circulation. – 2008. – Vol. 117. – Р. 526-535.
55. Besarab A., Bolton W.K., Browne J.K., et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin // N. Engl. J. Med. – 1998. – Vol. 339. – Р. 584-590.
56. Singh A.K., Szczech L., Tang K.L., et al. Correction of anemia with epoetin alfa in chronic kidney disease // N. Engl. J. Med. - 2006. - Vol. 355. - Р. 2085-98.
57. Pfeffer M.A., Burdman E.A., Chen C.Y., et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease // N. Engl. J. Med. – 2009. – Vol. 361. – Р. 2019-32.
58. Swedberg K., Young J.B., Anand I.S., et al. Treatment of anemia with darbepoetin alfa in systolic heart failure // N. Engl. J. Med. – 2013. – Vol. 368. – Р. 1210-9.
59. Jackevicius C.A., Shutieng Fan C., Warner A. Clinical outcomes of erythropoietin use in heart failure patients with anemia of chronic kidney disease // J. Cardiac. Failure. – 2014. – Vol. 20. – Р. 327-333.
60. Vaziri N.D., Zhou X.J. Potential mechanisms of adverse outcomes in trials of anemia correction with erythropoietin in chronic kidney disease // Nephrol. Dial. Transplant. – 2009. – Vol. 24. – P. 1082-8.
61. Kleijn L., Westenbrink B.D., von der Meer P. Erythropoietin and heart failure: the end of a promise? // Europ. J. Heart Failure. – 2013. – Vol. 15. – Р. 479-481.
62. Anker S.D., von Haehling S. Iron deficiency and anemia in heart failure, 2nd ed. Bremen, Germany: UNI-MED Verlag; 2012.
63. McDonagh T., Macdougall I.C. Iron therapy for the treatment of iron deficiency in chronic heart failure: intravenous or oral? // Europ. J. Heart Failure. – 2015. – Vol. 17. – Р. 248-262.
64. Crichton R.R., Danielson B.G., Geiser P. Iron therapy with special emphasis on intravenous administration, 4th ed. Bremen, Germany: UNI-MED Verlag AG; 2008.
65. Toblli J.E., Cao G., Olivieri L., et al. Comparative study of gastrointestinal tract and liver toxicity of ferrous sulfate, iron amino chelate and iron polymaltose complex in normal rats // Pharmacology. – 2008. – Vol. 82. – Р. 127-137.
66. Reinisch W., Staun M., Bhandari S., et al. State of the iron: how to diagnose and efficiently treat iron deficiency anemia in inflammatory bowel disease // J. Crohns. Colitis. – 2013. – Vol. 7. – P. 429-440.
67. Geisser P., Burkhardt S. The pharmacokinetics and pharmacodynamics of iron preperations // Pharmaceutics. – 2011. – Vol. 3. – P. 12-33.
68. Ericksen K., Ulvik R.J., Grimstad T., et al. Effects of ferrous sulphate and non-ionic iron-polymaltose complex on markers of oxidative tissue damage in patients with inflammatory bowel disease // Aliment. Pharmacol. Ther. – 2005. – Vol. 22. – P. 831-838.
69. Sandek A., Bauditz J., Swidsinski A., et al. Altered intestinal function in patients with chronic heart failure // J. Amer. Coll. Cardiol. – 2007. – Vol. 50. – P. 1561-1569.
70. Sandek A., Swidsinski A., Schroedil W., et al. Intestinal blood flow in patients with chronic heart failure. A link with bacterial growth, gastrointestinal symptoms, and cachexia // J. Amer. Coll. Cardiol. – 2014. – Vol. 64. – P. 1092-1102.
71. Darshan D., Frazer D.M., Anderson G.J. Molecular basis of iron-loading disorders // Expert. Rev. Mol. Med. – 2010. – Vol. 12. – e36.
72. Macdougall I.C. Iron supplementation in the non-dialysis kidney disease (ND-CKD) patient: oral or intravenous? // Curr. Med. Res. Opin. – 2010. – Vol. 26. – P. 473-482.
73. Nielsen P., Kongi R., Buggisch P., et al. Biovailability of oral iron drugs as judged by a 59Fe-whole-body counting technique in patients with iron deficiency anemia. Therapeutic efficacy of iron (II)-glycine sulfate // Arzneimittelforschung. – 2005. – Vol. 55. – P. 376-381.
74. Danielson B.G. Structure, chemistry, and pharmacokinetics of intravenous iron agents // J. Am. Soc. Nephrol. – 2004. – Vol. 15 (Suppl 2). – S93-S98.
75. Aapro M., Osterborg A., Gascon P., et al. Prevalence and management of cancer-related anemia, iron deficiency and the specific role of i. v. iron // Ann. Oncol. – 2015. – Vol. 23. – P. 1954-1962.
76. Bailie G.R., Clark J.A., Lane C.E., et al. Hypersensitivity reactions and deaths associated with intravenous iron preparations // Nephrol. Dial. Transplant. – 2005. – Vol. 20. – P. 1443-1449.
77. National Kidney Foundation. KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. 3.2. Using iron agents // Amer. J. Kidney Dis. – 2006. – Vol. 47 (Suppl 3). – S58-S70.
78. Geiser P., Baer M., Schaub E. Structure/histoxicity relationship of parenteral iron preparations // Arzneimittelforschung. – 1992. – Vol. 42. – P. 1439-1452.
79. McDonagh T., Macdougall I.C. Iron therapy for the treatment of iron deficiency in chronic heart failure: intravenous or oral? // Europ. J. Heart Failure. – 2015. – Vol. 17. – P. 248-262.
80. Tobili J.E., Lonbrana A., Gennaro F., et al. Intravenous iron reduces NT-pro-brain natrieretic peptide in anemic patients with chronic heart failure and renal insufficiency // J. Amer. Coll. Cardiol. – 2007. – Vol. 50. – P. 1657-1665.
81. Okonko D.O., Grzeslo A., Witkowski T., et al. Effect intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron deficiency. FERRIC-HF: a randomized, controlled, observer-blinded trial // J. Amer. Coll. Cardiol. – 2008. – Vol. 51. – P. 103-112.
82. Canzoni A.M. Intravenous iron-dextran: therapeutic and experiment possidilities // Schweiz Med. Wochensch. – 1970. – Vol. 100. – P. 301-3.
Журнал "Серцева недостатність та коморбідні стани" № 3, грудень 2017 р.
СТАТТІ ЗА ТЕМОЮ Кардіологія
Дисліпідемія та атеросклеротичні серцево-судинні захворювання (АСССЗ) є провідною причиною передчасної смерті в усьому світі (Bianconi V. et al., 2021). Гіперхолестеринемія – третій за поширеністю (після артеріальної гіпертензії та дієтологічних порушень) фактор кардіоваскулярного ризику в світі (Roth G.A. et al., 2020), а в низці європейських країн і, зокрема, в Польщі вона посідає перше місце. Актуальні дані свідчать, що 70% дорослого населення Польщі страждають на гіперхолестеринемію (Banach M. et al., 2023). Загалом дані Польщі як сусідньої східноєвропейської країни можна екстраполювати і на Україну....
Інколи саме з цього перерізу вдається візуалізувати тромбоемболи в основних гілках легеневої артерії або вегетації на стулках легеневого клапана (що трапляється надзвичайно рідко). Нахиливши датчик до самої верхівки серця, можна отримати її переріз по короткій осі, на якому, знову ж таки, порожнина лівого шлуночка має круглясту форму, а правого шлуночка – близьку до трикутника із виразною трабекулярністю (рис. 22.9). Розглядаючи зображення, також звертають увагу на те, що в нормі всі сегменти ЛШ скорочуються синхронно, не випереджаючи інші і не відстаючи. ...
Застосування статинів середньої інтенсивності в комбінації з езетимібом порівняно зі статинами високої інтенсивності окремо може забезпечити більше зниження рівня холестерину ліпопротеїнів низької щільності (ХС ЛПНЩ) у пацієнтів із нещодавнім ішемічним інсультом. Пропонуємо до вашої уваги огляд статті Keun-Sik Hong et al. «Moderate-Intensity Rosuvastatin Plus Ezetimibe Versus High-Intensity Rosuvastatin for Target Low-Density Lipoprotein Cholesterol Goal Achievement in Patients With Recent Ischemic Stroke: A Randomized Controlled Trial», опублікованої у виданні Journal of Stroke (2023; 25(2): 242‑250). ...
Артеріальна гіпертензія (АГ) сьогодні є одним із найпоширеніших серцево-судинних захворювань (ССЗ), що асоціюється з високим кардіоваскулярним ризиком, особливо в коморбідних пацієнтів. Навіть помірне підвищення артеріального тиску (АТ) пов’язане зі зменшенням очікуваної тривалості життя. До 40% хворих на АГ не підозрюють у себе недугу, бо це захворювання на початку може мати безсимптомний перебіг. Оптимальний контроль АТ є вагомим чинником профілактики фатальних серцево-судинних подій (ССП) для забезпечення якісного та повноцінного життя таких хворих. ...