



Трансформація гіпертрофічної кардіоміопатії в дилатаційну з подальшим зворотним розвитком
Вступ
Гіпертрофічна кардіоміопатія (ГКМП) є найчастішою причиною раптової смерті серед молодих пацієнтів. Разом із тим перебіг захворювання має варіабельний і гетерогенний характер. Вважають, що в пацієнтів із необструктивною формою ГКМП стан залишається стабільним тривалий час; рідко, в окремих хворих може розвинутися кінцева стадія серцевої недостатності (СН) з порушенням систолічної функції й перетворенням колишнього ГКМП-фенотипу на дилатаційний [1]. Обговорюють кілька механізмів, які лежать в основі такої трансформації, включаючи генетичні особливості та ушкодження, індуковані мікроваскулярною ішемією. Реверсійне ремоделювання належить до таких рідкісних випадків. На додаток тимчасове прогресування захворювання може провокуватися стрес-факторами, первинно незалежними від стану серця. Тому диференційна діагностика так званої кінцевої стадії ГКМП має важливе значення для вибору специфічних методів лікування.
Пропонуємо клінічний випадок молодої пацієнтки з ГКМП, у якої декомпенсована дилатаційна кардіоміопатія (ДКМП) була спричинена запаленням міокарда, а не первинно залежним від ГКМП процесом, і протизапальна терапія призвела до зворотного розвитку ремоделювання серця.
Клінічний випадок
Пацієнтка – 17-річна дівчина з діагнозом ГКМП без обструкції вихідного тракту лівого шлуночка (ЛШ), із концентричною гіпертрофією (передньо-септальна стінка – 15 мм, задня – 16 мм), зі збереженою фракцією викиду (ФВ) та синдромом Wolff-Parkinson-White (WPW), з приводу якого в дитинстві двічі проводили абляцію.
Була госпіталізована для обстеження з приводу прогресування СН, що проявлялася вперше діагностованим зниженням ФВ до 40%, гіпокінезією верхівки та нижньої стінки, збільшенням розмірів ЛШ (кінцево-діастолічний розмір (КДР) – 59 мм, мітральний клапан-перегородка – 18 мм), підвищенням тиску наповнення та мітральною регургітацією ІІ. Під час холтерівського моніторування електрокардіограми зареєстровано численні епізоди шлуночкових і суправентрикулярних порушень ритму. Клінічно хвора скаржилася на біль у грудях, задишку, у тому числі в стані спокою (III функціональний клас за NYHA), напади серцебиття з пресинкопальними епізодами протягом минулих 6 міс. Аналіз крові показав підвищення концентрації мозкового натрійуретичного пропептиду (МНУпП) до 5201 пг/мл та незначне зростання рівнів трансаміназ (АЛТ – 40 Од/л, АСТ – 60 Од/л) без змін рівнів тропоніну Т, С-реактивного протеїну (СРП) і кількості лейкоцитів. Протягом 7-денного періоду спостереження ФВ ЛШ знизилася до 20%, вказуючи на швидке прогресування СН.
Ураховуючи результати обстеження, обговорювали щонайменше 4 можливі діагнози:
1) кардіоміопатія, індукована тахікардією;
2) прогресування ГКМП через зміни, спричинені мікроангіопатією;
3) розвиток тяжкої первинної мітральної регургітації;
4) інша невстановлена набута форма кардіоміопатії.
Під час інвазивного електрофізіологічного дослідження не вдалося спровокувати злоякісні порушення серцевого ритму. Атріовентрикулярний вузол функціонував адекватно без будь-яких ознак додаткових провідних шляхів, що виключило можливість тахікардіоміопатії, пов’язаної з WPW-синдромом.
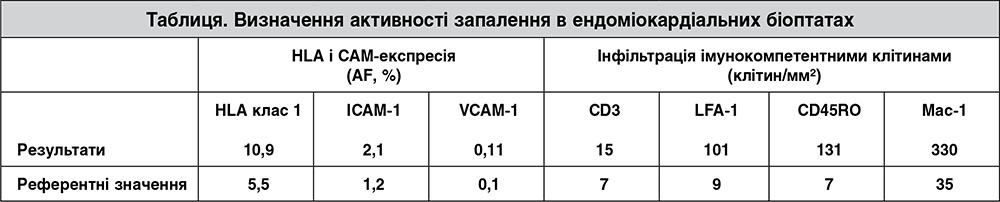
Трансезофагальна ехокардіографія виключила первинну недостатність мітрального клапана. Для подальшого дослідження структури міокарда хворій виконали магнітно-резонансну томографію (МРТ), яка продемонструвала значну гіпертрофію й дилатацію ЛШ (кінцево-діастолічний об’єм ЛШ – 286 мл) із подальшим зниженням ФВ (17%). Також визначалися ділянки посиленого МР-сигналу на пізніх післяконтрастних зображеннях, що свідчило на користь некротичних (рубцевих) та/або фібротичних змін, особливо в міжшлуночковій перетинці, у ділянці верхівки та латеральній ділянці ЛШ (ретикулярне відтерміноване накопичення) (рис. 1). При цьому не було ознак запального процесу в міокарді на Т1- і Т2-зважених зображеннях. Для того щоб визначити, чи пов’язане прогресування захворювання з ГКМП або воно має іншу етіологію, пацієнтці після виключення ураження коронарних судин виконали ендоміокардіальну біопсію (ЕМБ) правого шлуночка. Гістологічна картина ЕМБ продемонструвала периваскулярний та інтерстиціальний фіброз і значну гіпертрофію кардіоміоцитів із діаметром до 31 мкм.
Типового для ГКМП феномена «disarray» (із невпорядкованим розташуванням м’язових волокон) виявлено не було. Також не знайшли ознак хвороб накопичення після використання різних методик фарбування гістологічних препаратів. Проте імуногістохімічне дослідження показало наявність поширеного активного запального процесу в міокарді (рис. 2): значне збільшення β2-лейкоцит-інтегринів/інфільтратів (LFA‑1/CD11a+ та Mac‑1/CD11b+) і змішаних клітинних інфільтратів із лімфоцитів і макрофагів (CD45RO-позитивні та HLA-позитивні клітини) (табл.) [2]. Молекулярно-біологічний аналіз за допомогою полімеразно-ланцюгової реакції не виявив кардіотропних вірусів, включаючи ентеровірус, аденовірус, вірус Epstein-Barr, людський вірус герпесу 6 і парвовірус В19 [3].
Спираючись на результати ЕМБ, які підтвердили наявність вірус-негативного активного міокардиту, що, найімовірніше, й зумовив порушення функції серця, хворій призначили імуносупресивну терапію з кортикостероїдами (преднізолон у дозі 1 мг/кг/добу) та азатіоприном (100 мг/добу) під контролем функції печінки/нирок і з регулярним підрахунком формули крові. Через місяць дозу преднізолону почали зменшувати на 10 мг що 4 тиж, поки не досягли підтримувальної дози – 10 мг/добу [2]. Упродовж 3 міс після початку лікування ФВ ЛШ поступово підвищилася до 52%, а КДР зменшився до 56 мм. Рівень МНУпП знизився до 968 пг/мл, показники трансаміназ, СРП та кількість лейкоцитів – у межах норми. На додаток клінічний стан пацієнтки також покращився.
Підсумовуючи, автори зазначають, що зовнішні фактори, в цьому випадку міокардит, можуть спровокувати перетворення ГКМП-фенотипу на ДКМП-фенотип. Специфічна протизапальна терапія після ЕМБ-діагностики здатна забезпечити зворотний розвиток захворювання.
Дискусія
Зазвичай розвиток ГКМП зумовлений генетичними розладами: приблизно в 40-60% випадків причиною захворювання є мутації генів, які кодують протеїни кардіосаркомерів (міозин-зв’язувальний протеїн С, важкі ланцюги β-міозину, серцеві тропоніни І і Т, легкі ланцюги міозину III та α‑1 ланцюги тропоміозину).
Особливо це стосується дітей і підлітків з ізольованою гіпертрофією серця, понад половина всіх таких випадків є генетично детермінованими [4]. Крім того, відомі змішані генетичні порушення, що спричиняють ГКМП і порушення ритму, так званий PRKAG2-кардіальний синдром, під яким розуміють наявність ГКМП, шлуночкової преекзитації й тахіаритмії (WPW-синдром), а також прогресуюче захворювання провідної системи серця [5]. Такі генетичні розлади вперше були описані в канадської сім’ї французького походження на прізвище Gollob [6], вони пов’язані з мутацією гена PRKAG2, що кодує γ2-субодиницю αβγ-гетеротримеру АМФ-активуючої протеїнкінази [7-10]. Проте PRKAG2-синдром у нашої пацієнтки був виключений іще за попередніх обстежень. Окрім того, аналіз інших відомих мутацій генів MYH7, MYBPC3, TNNT2, TPM1, TNNI3, MYL3, MYL2, CSRP3, PLN, ACTC і TNNC1 не привів у неї до певних результатів. Загальновизнано, що в аж до 30% випадків ГКМП специфічних мутацій не знаходять або вони є досі невідомими. Нарешті, до 20% пацієнтів, які мають значну гіпертрофію ЛШ, не належать до класичних форм ГКМП, але мають інші генетичні й негенетичні причини, у тому числі хвороби накопичення [11, 12].
Під впливом стресових факторів існує можливість переходу ГКМП у дилатаційний фенотип, але це трапляється не дуже часто й головним чином за тяжких форм ГКМП з обструкцією ЛШ. До додаткових зовнішніх стресових факторів належать емоційний стрес, токсичні субстанції, такі як алкоголь або хіміотерапія, вагітність, ішемія, індуковані тахікардією порушення серцевого ритму та запалення. За даними Matsumori і співавт. до таких факторів ризику може належати й вірусний міокардит [13]. Сам по собі вірус у міокарді, як і запальні реакції, здатний погіршити перебіг ГКМП, що разом із підвищенням інтравентрикулярного тиску може призвести до дилатації ЛШ [14]. Цей механізм, описаний у пацієнтів із ГКМП та міокардитом, імовірно, причетний до погіршення функції серця й розвитку електричної нестабільності [15].
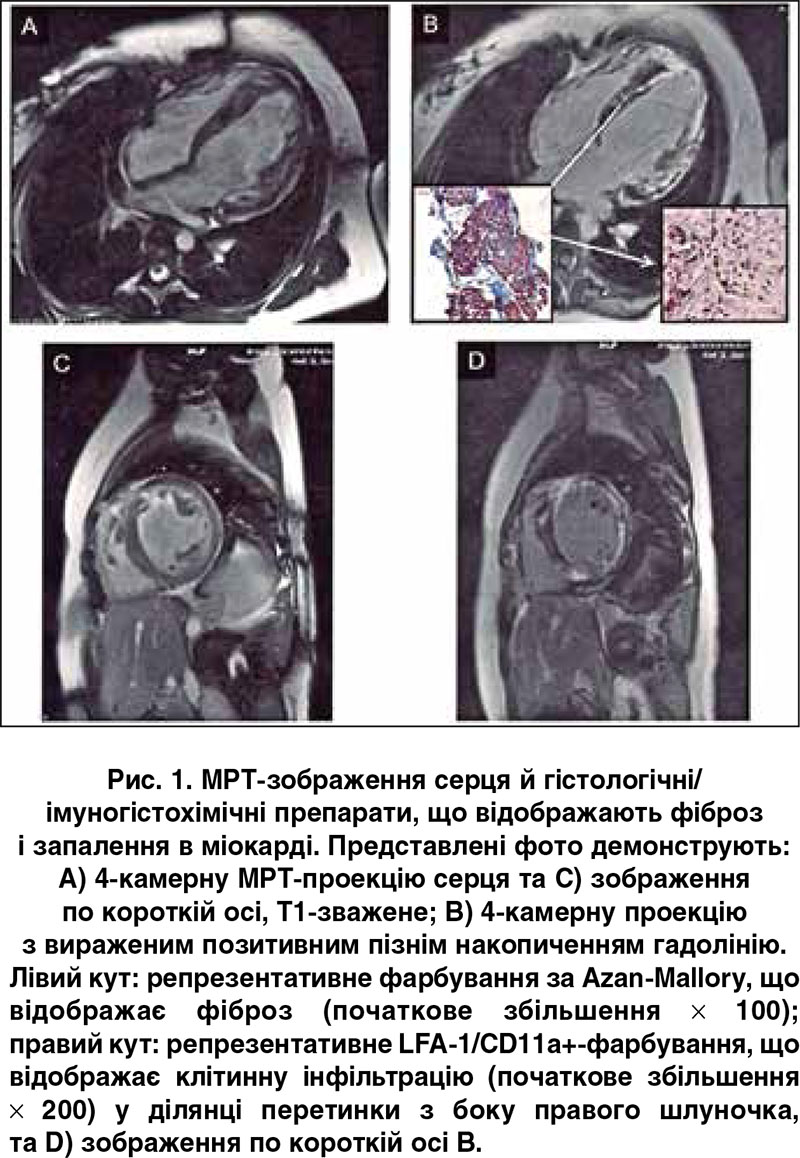
Зважаючи на результати ЕМБ, ми дійшли висновку, що пацієнтка мала вірус-негативний активний міокардит. Імуногістохімічний аналіз підтвердив значну, частково зливну лімфоцитарну інфільтрацію й підвищену експресію молекул адгезії. Він також виявив помітні інфільтрати з макрофагів та інших характерних для запалення клітин, але не було знайдено специфічних цитотоксичних Т-лімфоцитів (перфорин-позитивних). На противагу біопсії, під час МРТ специфічні ознаки міокардиту не були виявлені, але знайдені ділянки фіброзу (LGE), розвиток якого можна пояснити основним захворюванням або додатковим ушкодженням за рахунок запального процесу в міокарді. Наявність фіброзу також відзначали й у біоптатах, узятих із LGE-ділянок перетинки (рис. 1B). Проте тільки наступне цитологічне дослідження цих біоптатів (із фарбуванням характерних для запалення клітин) дало змогу діагностувати тяжкий вірус-негативний активний міокардит (рис. 2). Отримані результати узгоджуються з даними інших дослідників, які свідчать, що інформативність МРТ щодо виявлення запального процесу (чутливість – 76%, специфічність – 54% і точність – 68%) має істотні обмеження й може бути недостатньою для діагностики міокардиту в усіх випадках [16]. Негативні результати МРТ не виключають автоматично наявність міокардиту й потребують подальшого дослідження з проведенням ЕМБ, як зазначено робочою групою Європейського товариства кардіологів у рекомендаціях щодо міокардиту та хвороб перикарда [17].
Діагностика тяжкого вірус-негативного активного міокардиту за результатами ЕМБ дала нам змогу негайно розпочати імуносупресивну терапію, що включала преднізолон та азатіоприн протягом 6 міс, як радять Caforio і співавт. [17]. Як і в дослідженні TIMIC [18], ми спостерігали відновлення ФВ ЛШ і зменшення КДР ЛШ уже після 3 міс лікування, що супроводжувалося покращенням клінічного стану й еволюцією з ІІІ до І функціонального класу за класифікацією NYHA.
У підсумку цей випадок показує, що міокардит може сприяти трансформації гіпертрофічного фенотипу в дилатаційний, із подальшим зворотним розвитком ремоделювання міокарда після початку спеціальної протизапальної терапії.
Література
1. Harris K.M., Spirito P., Maron M.S., Zenovich A.G., Formisano F., Lesser J.R., Mackey-Bojack S., Manning W.J., Udelson J.E., Maron B.J. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation, 2006; 114: 216-225.
2. Noutsias M., Pauschinger M., Schultheiss H.P., Kuhl U. Phenotypic characterization of infiltrates in dilated cardiomyopathy-diagnostic significance of T-lymphocytes and macrophages in inflammatory cardiomyopathy. Med. Sci. Monit. 2002; 8: CR478-CR487.
3. Kuhl U., Pauschinger M., Noutsias M., Seeberg B., Bock T., Lassner D., Poller W., Kandolf R., Schultheiss H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation, 2005; 111: 887-893.
4. Morita H., Rehm H.L., Menesses A., McDonough B., Roberts A.E., Kucherlapati R., Towbin J.A., Seidman J.G., Seidman C.E. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 2008; 358: 1899-1908.
5. Cherry J.M., Green M.S. Familial cardiomyopathy: a new autosomal dominant form (abstract). Clin. Invest. Med. 1986; 9: B31.
6. Gollob M.H. Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem. Soc. Trans. 2003; 31 (Pt. 1): 228-231.
7. Wolf C.M., Arad M., Ahmad F., Sanbe A., Bernstein S.A., Toka O., Konno T., Morley G., Robbins J., Seidman J.G., Seidman C.E., Berul C.I. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation, 2008; 117: 144-154.
8. Akman H.O., Sampayo J.N., Ross F.A., Scott J.W., Wilson G., Benson L., Bruno C., Shanske S., Hardie D.G., Dimauro S. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr. Res. 2007; 62: 499-504.
9. Burwinkel B., Scott J.W., Buhrer C., van Landeghem F.K., Cox G.F., Wilson C.M., Hardie D.G., Kilimann M.W. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am. J. Hum. Genet. 2005; 76: 1034-1049.
10. Hendrickx J., Lee P., Keating J., Carton D., Sardharwalla I.B., Tuchman M., Baussan C., Willems P.J. Complete genomic structure and mutational spectrum of PHKA2 in patients with X-linked liver glycogenosis type I and II. Am. J. Hum. Genet. 1999; 64: 1541-1549.
11. Elliott P.M., Anastasakis A., Borger M.A., Borggrefe M., Cecchi F., Charron P., Hagege A.A., Lafont A., Limongelli G., Mahrholdt H., McKenna W.J., Mogensen J., Nihoyannopoulos P., Nistri S., Pieper P.G., Pieske B., Rapezzi C., Rutten F.H., Tillmanns C., Watkins H., Authors/Task Force members. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the element of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014; 35: 2733-2779.
12. Arad M., Maron B.J., Gorham J.M., Johnson W.H. Jr., Saul J.P., Perez-Atayde A.R., Spirito P., Wright G.B., Kanter R.J., Seidman C.E., Seidman J.G. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005; 352: 362-372.
13. Matsumori A., Matoba Y., Nishio R., Shioi T., Ono K., Sasayama S. Detection of hepatitis C virus RNA from the heart of patients with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 1996; 222: 678-682.
14. Frustaci A., Verardo R., Caldarulo M., Acconcia M.C., Russo M.A., Chimenti C. Myocarditis in hypertrophic cardiomyopathy patients presenting acute clinical deterioration. Eur. Heart J. 2007; 28: 733-740.
15. Chimenti C., Calabrese F., Thiene G., Pieroni M., Maseri A., Frustaci A. Inflammatory left ventricular microaneurysms as a cause of apparently idiopathic ventricular tachyarrhythmias. Circulation, 2001; 104: 168-173.
16. Lurz P., Eitel I., Adam J., Steiner J., Grothoff M., Desch S., Fuernau G., de Waha S., Sareban M., Luecke C., Klingel K., Kandolf R., Schuler G., Gutberlet M., Thiele H. Diagnostic performance of CMR imaging compared with EMB in patients with suspected myocarditis. JACC Cardiovasc. Imaging. 2012; 5: 513-524.
17. Caforio A.L., Pankuweit S., Arbustini E., Basso C., Gimeno-Blanes J., Felix S.B., Fu M., Helio T., Heymans S., Jahns R., Klingel K., Linhart A., Maisch B., McKenna W., Mogensen J., Pinto Y.M., Ristic A., Schultheiss H.P., Seggewiss H., Tavazzi L., Thiene G., Yilmaz A., Charron P., Elliott P.M., European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013; 34: 2636-2648; 2648a‑2648d.
18. Frustaci A., Russo M.A., Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur. Heart J. 2009; 30: 1995-2002.
Переклала з англ. Ганна Лях
СТАТТІ ЗА ТЕМОЮ Кардіологія
Як відомо, кальцій бере участь у низці життєво важливих функцій. Хоча більшість досліджень добавок кальцію фокусувалися переважно на стані кісткової тканини та профілактиці остеопорозу, сприятливий вплив цього мінералу є значно ширшим і включає протидію артеріальній гіпертензії (передусім у осіб молодого віку, вагітних та потомства матерів, які приймали достатню кількість кальцію під час вагітності), профілактику колоректальних аденом, зниження вмісту холестерину тощо (Cormick G., Belizan J.M., 2019)....
Торакалгія – симптом, пов’язаний із захворюваннями хребта. Проте біль у грудній клітці може зустрічатися за багатьох інших захворювань, тому лікарям загальної практики важливо проводити ретельну диференційну діагностику цього патологічного стану та своєчасно визначати, в яких випадках торакалгії необхідна консультація невролога. В березні відбувся семінар «Академія сімейного лікаря. Біль у грудній клітці. Алгоритм дій сімейного лікаря та перенаправлення до профільного спеціаліста». Слово мала завідувачка кафедри неврології Харківського національного медичного університету, доктор медичних наук, професор Олена Леонідівна Товажнянська з доповіддю «Торакалгія. Коли потрібен невролог»....
Рівень ліпопротеїну (a) >50 мг/дл спостерігається в ≈20-25% населення і пов’язаний із підвищеним ризиком серцево-судинних захворювань (ССЗ) [1]. Ліпопротеїн (a) задіяний в атерогенезі та судинному запаленні, а також може відігравати певну роль у тромбозі через антифібринолітичну дію і взаємодію із тромбоцитами [2, 3]. Дієта та фізична активність не впливають на рівень ліпопротеїну (a); специфічної терапії для його зниження також не існує. Підвищений ризик ССЗ, пов’язаний з ліпопротеїном (а), залишається навіть у пацієнтів, які приймають статини [4]. Саме тому існує критична потреба в терапії для зниження цього ризику, особливо в первинній профілактиці. ...
Запалення відіграє важливу роль у розвитку багатьох хронічних захворювань, зокрема атеросклерозу. Нещодавно було встановлено, що гіперурикемія спричиняє запалення ендотеліальних клітин судин, ендотеліальну дисфункцію та, зрештою, атеросклероз. Експериментальна робота Mizuno та співавт. (2019), у якій було продемонстровано здатність фебуксостату пригнічувати запальні цитокіни, привернула увагу дослідників до протизапальних ефектів уратзнижувальних препаратів. Кількість лейкоцитів – надійний маркер запалення, пов’язаний із різними кардіоваскулярними захворюваннями, як-от ішемічна хвороба серця; у багатьох попередніх дослідженнях його використовували для оцінки протизапального ефекту терапевтичного втручання. Мета нового аналізу дослідження PRIZE – вивчити вплив фебуксостату на кількість лейкоцитів у пацієнтів із безсимптомною гіперурикемією....