



Печінка та метаболізм заліза: комплексна гіпотеза патогенезу спадкового гемохроматозу
 Е.Г. Манжалій*
Е.Г. Манжалій*Фізіологія метаболізму заліза
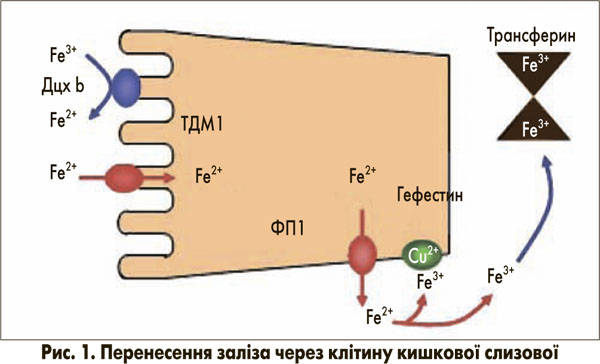
Для підтримання нормального рівня гемоглобіну вміст заліза повинен дозволяти підвищувати поглинання заліза в разі залізодефіцитної анемії. Вважається, що за фізіологічних умов втрата заліза організмом виникає тільки при пасивній десквамації клітин, особливо клітин кишкового епітелію. Як правило, постійні втрати становлять приблизно 1 мг на день. Відповідно, контроль запасів заліза може бути опосередкований лише шляхом регуляції процесу всмоктування. Засвоєння їжі в кишечнику відповідає потребам організму для підтримання балансу заліза. У молекулярному узгодженні процесу поглинання клітинами слизової дванадцятипалої кишки беруть участь декілька білків, пов’язаних із мембраною, більшість із яких добре вивчені (рис. 1) [1]. На люмінальній поверхні редуктаза заліза дуоденального цитохрому b (Дцх b) сприяє перетворенню Fe3+ на Fe2+, що є єдиною формою заліза, яка може проходити крізь біологічні мембрани. Перенесення Fe2+ через апікальну плазматичну мембрану регулює ТДМ1 (iзоформа 1) [2], який не є селективним для Fe2+ і також переносить інші двовалентні метали. Внутрішньоклітинний пасаж до кінця незрозумілий і може залучати цитозольні транспортні білки, такі як феритин або фізіологічні комплексони, які зв’язують залізо при нетоксичному стані. На базолатеральній мембрані феропортин 1 (ФП1) транспортує Fe2+ через мембрану. Гефестин (церулоплазмін-подібний протеїн), розташований на зовнішньому шарі клітинної мембрани, окислює Fe2+ до Fe3+, а отже, забезпечує його зв’язок із трансферином, головним транспортним білком заліза в крові. Трансферин доставляє залізо до місць його метаболізму – переважно еритропоетинової системи та печінки. Тут залізо захоплюється трансфериновим рецептором 1 типу ендоцитозного шляху. Вивільнення із закисленого внутрішньоклітинного компартмента в цитозоль відбувається за допомогою ТДМ1 (ізоформа 2) [2].
Залізо необхідне всім клітинам для включення в залізовмісні білки. Його наявність є найбільш важливою в еритропоетиновій системі, де доставка заліза безпосередньо відповідає потребам для продукції гемоглобіну [3]. Більшість заліза, необхідного для еритрону, використовується повторно з макрофагів РЕС, що беруть метал зі старих еритроцитів (рис. 2) [3]. У клітинах РЕС залізо, що походить із гемоглобіну, переноситься від фагосом у цитозоль ТДМ1 (ізоформа 2). Та, як описано для кишкової слизової, вивільняється з цих клітин шляхом ФП1.
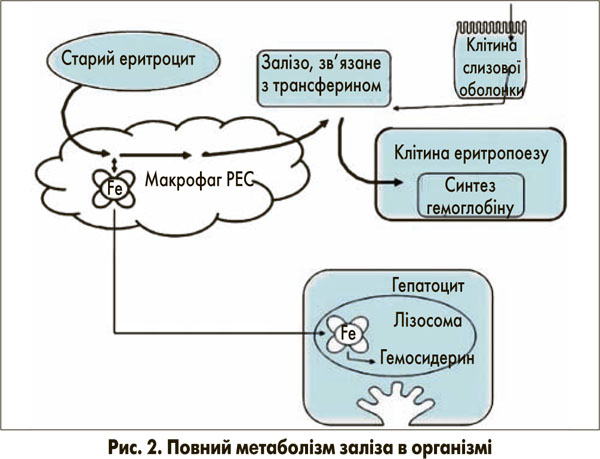
На зовнішньому шарі плазматичної мембрани Fe2+ перетворюється на Fe3+ за допомогою церулоплазміну та знову сполучається з трансферином для доставки до місць потреби, наприклад, до клітин еритропоезу. Надлишкове залізо, що виникає при розпаді еритроцитів та гемолізі або при підвищеному всмоктуванні при гемохроматозі, сполучається з феритином за допомогою макрофагів РЕС. Феритин – це Fe3+-концентруючий протеїн, який за своєю природою захищає клітини від надмірної кількості незв’язаного заліза та, відповідно, від пошкодження вільними радикалами. Вважається, що в разі потреби залізо може вивільнятися з феритину. При надлишку заліза, однак, феритин, навантажений залізом, вивільняється в кровоток та швидко виводиться з нього за допомогою ще до кінця не з’ясованих механізмів. Можливо, у них залучені зумовлені феритиновим рецептором процеси захоплення в гепатоцитах із подальшою лізосомальною секвестрацією. Яким чином залізо може бути вивільнене з гепатоцитів у жовч, потрібно далі вивчати. На ефективний гепатоцелюлярний кліренс феритину не впливає вміст заліза в печінці, це може бути причиною пояснювати те, чому печінка містить відносно високий рівень заліза, яке не метаболізується.
Регуляція метаболізму заліза
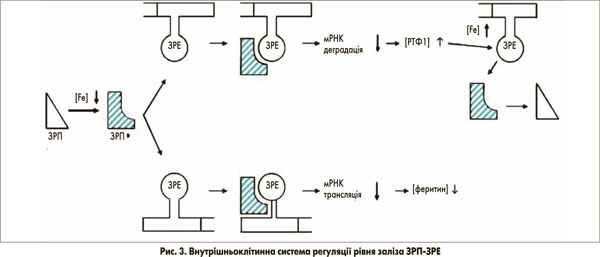
Засвоєння заліза, зв’язаного з трансферином через РТФ1, так само, як і відкладення у феритині, лежить в основі посттранскрипційного механізму контролю. Він залучає внутрішньоклітинні залізорегулюючі білки (ЗРБ1 та 2), які зв’язуються зі специфічною стовбурово-петлевою структурою (залізозв’язуючий елемент – ЗЗЕ) нетрансльованої ділянки мРНК ключових залізовмісних білків, наприклад, РТФ1 та феритину (рис. 3) [5]. ЗРБ1 активується, коли в клітин виникає потреба в залізі. Зв’язуючись з 3’-кінцем залізозв’язуючого елемента, мРНК РТФ1 інгібує її руйнування РНКазами та послідовно стабілізує мРНК. У результаті клітинне накопичення РТФ1 компенсує низький вміст заліза в клітині. Цікаво, що ТДМ1 (ізоформа 1), який міститься в клітинній мембрані епітеліальних клітин і відповідає за клітинне поглинання заліза, не зв’язаного з феритином, також містить 3’-залізозв’язуючий елемент [6]. Для порівняння, ізоформа 2, якої особливо багато в еритроїдних клітинах-попередниках і яка відповідає за ендосомальне вивільнення заліза, не має ЗЗЕ. Одночасне зв’язування ЗРБ1 з 5’-кінцем залізозв’язуючого елемента не трансльованої мРНК феритину запобігає транскрипції цього запасного протеїну та будь-якому клітинному накопиченню заліза в подальшому, що є іншою допоміжною ланкою при компенсації клітинного дефіциту заліза.
Ми вважаємо, що існує захисний механізм, який викликає залізодефіцит при запаленні. Однак цей механізм сьогодні не можна назвати цілком зрозумілим. З одного боку, він характеризується низьким рівнем сироваткового заліза (низьке насичення трансферину залізом), а з іншого – високим сироватковим рівнем феритину. Це свідчить про те, що всмоктування заліза знижене і будь-який надлишок сироваткового заліза одразу потрапляє у феритинову «пастку». Сповільнення всмоктування заліза викликане зниженням рівня ФП1-помпи, що перекачує залізо, наявне в клітинах кишкової слизової. ФП1 також у великій кількості присутній у клітинах РЕС. Отже, типовий шлях блокування ФП1 при запаленні призводить до зниженого вивільнення заліза з цих двох типів клітин і подальшої внутрішньоклітинної секвестрації заліза у формі феритину. З макрофагів РЕС феритин вивільняється в сироватку та сприяє гіперферитинемії, що спостерігається при запаленні. Водночас насичення трансферину є низьким через знижене всмоктування заліза клітинами слизової оболонки дванадцятипалої кишки. Анемія при хронічному запаленні може бути результатом цього залізозбіднюючого механізму [7].
Ключовим у цьому процесі є гепсидин, залізо-регулюючий «гормон». Він переважно синтезується в гепатоцитах. Експресія гепсидину регулюється запальними сигналами, наприклад, інтерлейкіном‑6, що призводить до підвищеного виділення його в кровоток, тимчасом як залізодефіцит призводить до пригнічення синтезу гепсидину [8, 9].
Було неочікувано, що рецептором гепсидину виявився ФП1, який присутній у клітинах слизових оболонок, РЕС, а також в клітинах плаценти та в меншій кількості – у гепатоцитах [10].
Зв’язування гепсидину з ФП1 призводить до об’єднання та деградації ФП1. Наслідком є дефіцит клітинного виходу вільного заліза та його зберігання у феритиновій «пастці». Таким чином, за високого рівня гепсидину залізо після всмоктування клітинами слизової оболонки не переноситься через базолатеральну клітинну мембрану. Таке пригнічення всмоктування заліза викликає залізодефіцит (і в результаті – низьке насичення трансферину залізом).
Транскрипційний контроль синтезу гепсидину виникає через вище розташовані перетворювачі гемоювелін (ГЮВ) та РТФ2 [11]. Ці білки містяться в клітинній мембрані та пов’язані з сигнальними шляхами для контролю транскрипції гепсидину (рис. 4).
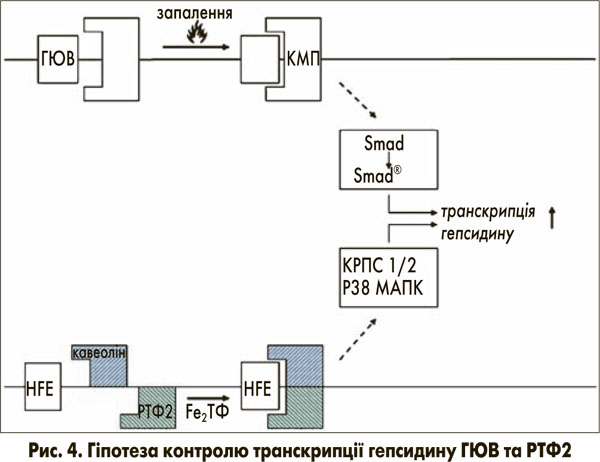
Гемоювелін відповідає на запальні медіатори дією, схожою на корецептор кісткового морфогенетичного протеїну (КМП), що призводить до фосфорилювання родини рецептор-активованого фактора транскрипції R-smad. Після зв’язування корецептора та smad, R-smad комплекс переноситься в ядро, де він потім посилює транскрипцію мРНК гепсидину [12].
РТФ2 вважається детектором рівня заліза в організмі (кількість заліза, зв’язаного з трансферином / насичення трансферину) [13]. РТФ2 зв’язує (Fe)2-ТФ і перетворює інформацію про рівень заліза в організмі на сигнал комплексу з ефектором з послідовною індукцією синтезу гепсидину [14]. Цікаво відмітити, що ген гемохроматозу зв’язується з РТФ2 для передачі сигналів про рівень заліза до клітини [14]. Більше того, нещодавні спостереження дають підстави припускати, що мембранний глікопротеїн РТФ2, локалізований у кавеолярних мікропросторах, взаємодіє з CD81 та кавеоліном 1 [15]. Цей комплекс активує кінази, що регулюються позаклітинними сигналами 1 та 2 (КРПС 1 та 2), та p38 мітоген-активовані протеїнкінази (МАПК), які, у свою чергу, контролюють синтез гепсидину (рис. 4). Ця гіпотеза може пояснити подвійну регуляцію синтезу гепсидину залізом через РТФ2 та запальні сигнали через ГЮВ.
Патогенетична концепція спадкового гемохроматозу
При патологічному стані спадкового гемохроматозу помірне, але постійно зростаюче базальне всмоктування заліза призводить до посилення навантаження на організм залізом до 20-40 г (нормальний вміст заліза в організмі – 5 г) у пацієнтів віком 40-60 років. Таке накопичення заліза зазвичай відбувається в гепатоцитах, а не в макрофагах, що в більшості випадків викликано гомозиготною мутацією C282Y гена гемохроматозу. Досі не до кінця зрозуміло, яким чином ця мутація призводить до підвищення всмоктування заліза. Нормальний HFE-білок зв’язується в ендоплазматичному ретикулумі з β2-мікроглобуліном і потім переноситься до клітинної мембрани (рис. 5), де він вбудовується в мембрану та з’єднується з РТФ1. Ми припускаємо, що транспортер двовалентних металів ТДМ1 (ізоформа 2) також зв’язується з цим комплексом для сприяння клітинному поглинанню заліза, зв’язаного з трансферином. Фактично в останніх дослідженнях показано, що ген гемохроматозу копреципітує з ТДМ1, вважається, що вони мають тісну внутрішньоклітинну взаємодію [16].
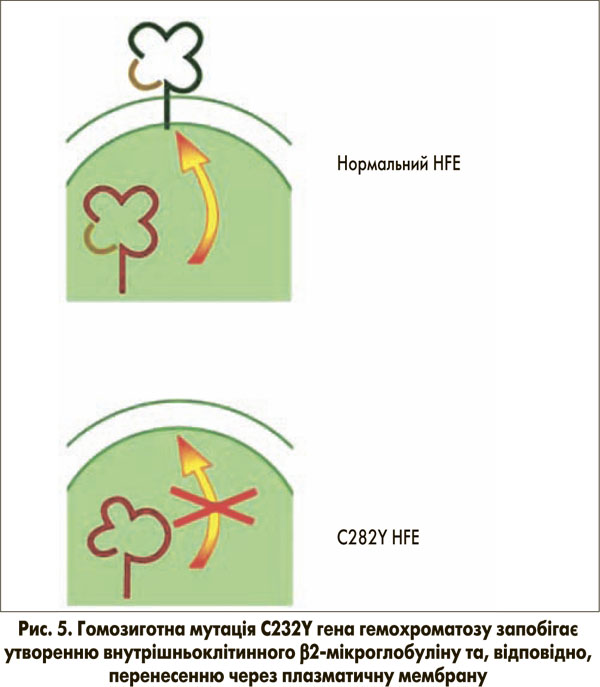
Мутація C282Y запобігає розміщенню гена гемохроматозу на клітинній мембрані (рис. 5). Як наслідок, утворення трансферинового комплексу поглинання HFE-РТФ1-ТДМ1 порушується. При цьому навантажений залізом трансферин все ще здатний зв’язуватися з РТФ1шляхом ендоцитозу. За відсутності гена гемохроматозу, однак, ТДМ1 може лише випадково зв’язуватися з комплексом (Fe)2-ТФ-РТФ1, а отже, залізо не може ефективно вивільнятися з заглиблень ендоцитозних пухирців. Таким чином, поглинання заліза в цитозолі порушене. Це може бути неважливим для системи еритропоезу через високий вміст РТФ1 та ізоформи 2 ТДМ1 (за відсутності гена гемохроматозу), але цей факт є істотним для гепатоцитів з відносно низьким рівнем експресії РТФ1. Порушене вивільнення заліза з ендосомальних везикул через порушений зв’язок РТФ1-ТДМ1 може бути причиною того, що залізо залишається зв’язаним із трансферином. Як наслідок, у гепатоцитах може виникати стан залізодефіциту (рис. 6). Це, у свою чергу, може пригнічувати синтез гепсидину для компенсації наявного залізодефіциту через механізми, описані вище.
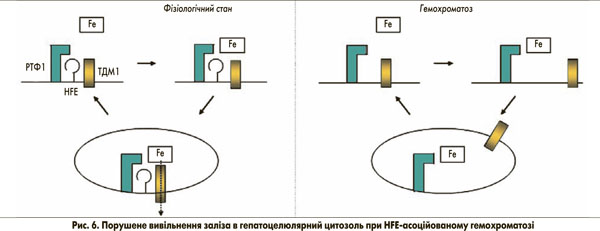
Із часом насичення трансферину залізом зростає, що є ключовою діагностичною ознакою при HFE-асоційованому гемохроматозі. Це пояснюється постійною циркуляцією заліза, зв’язаного з трансферином через ендосомальний компартмент і відповідним дефіцитом транспортування в цитозоль за допомогою ТДМ1.
Однак зворотній зв’язок інтенсивно насиченого трансферину з РТФ2 не може бути ефективним через дефіцит гена гемохроматозу (рис. 4) [14]. Таким чином, транскрипція гепсидину пригнічується і далі.
Кишкове всмоктування заліза підвищується протягом тривалого періоду, розвивається збільшення запасів заліза. Через низький рівень гепсидину та відповідну високу активність РТФ1 клітини РЕС не беруть участі в навантаженні залізом при спадковому гемохроматозі.
Постає питання: як перенавантаження залізом виникає в гепатоцитах за таких умов, коли РТФ1 також активний? Хоча точної відповіді немає, очевидно, що залізо, всмоктане в надмірній кількості, не утилізується еритроном для додаткового синтезу гемоглобіну. Отже, воно має сполучатися з феритином для зберігання, наприклад, клітинами РЕС. Цей феритин з інертним залізом, що не підлягає метаболізму, вивільняється в кровоток і захоплюється гепатоцитами для утилізації. Оскільки організм не розцінює це залізо як надмірне внутрішньоклітинне навантаження, то сигнал про низький рівень гепатоцелюлярного заліза, викликаний мутацією гена гемохроматозу, зберігається.
Пригнічена печінкова екскреція гепсидину підтримує сигнал до підвищеного всмоктування заліза. Під час перебігу хвороби рівень феритину в крові постійно зростає, призводячи до гіперферитинемії, іншої ключової ознаки спадкового гемохроматозу. Гепатоцити накопичують надмірний феритин, що надходить у лізосомальний компартмент, де він ущільнюється і може бути розщеплений до гемосидерину. Звідти вільне залізо може вийти та викликати пошкодження мембран вільними радикалами, що спричинить пошкодження клітин. Це, як і інше супутнє ураження (наприклад, алкогольне), викликає запальну відповідь, і рівень гепсидину зростає через гемоювеліновий шлях [12], врівноважуючи підвищене всмоктування заліза. Водночас гепатоцелюлярний ФП1 (клітинний насос перекачування заліза) «зачиняється», що погіршує вільнорадикальне пошкодження. Тривале гепатоцелюлярне пошкодження викликає загибель клітин, за якою слідує печінкова регенерація, що може призвести як до фіброзу та цирозу, так і сприяти гепатоканцерогенезу.
Поточне лікування HFE-асоційованого спадкового гемохроматозу шляхом флеботомій зменшує навантаження організму залізом. Отже, продукція феритину для зберігання надмірного заліза припиняється і паралельно падає сироватковий рівень феритину. Залізо тепер більш потрібне для синтезу гемоглобіну еритроном. Звісно, саме високі потреби еритрону в залізі спрямовують абсорбоване залізо, зв’язане з трансферином, на синтез гемоглобіну та запобігають відкладенню у феритині. Висока потреба еритрону в залізі також може бути причиною того, що жінки у період менструації рідко мають виражене перенавантаження залізом. Більше того, індивідуальні відмінності у потребі заліза можуть пояснити різну клінічну пенетрантність HFE-асоційованого гемохроматозу. Генетично-сприйнятливі особи (гомозиготи з мутацією C282Y) з низькою потребою в залізі схильні до перевантаження залізом, тимчасом як із високою потребою – ні.
HFE-неасоційований гемохроматоз
HFE-неасоційований гемохроматоз є виключно рідкісним і викликаний мутацією генів ГЮВ, РТФ2 або гепсидину (HAMP). Швидкість всмоктування заліза значно вища в порівнянні з HFE-асоційованим гемохроматозом, що призводить до більш вираженого перевантаження залізом [17, 18]. Очевидно, що пригнічення рівня гепсидину більш виражене у випадку HAMP-мутацій. Мутації генів ГЮВ та РТФ2 також спричиняють дуже низький рівень гепсидину, оскільки вони є безпосередньо зв’язаними вищими регуляторами синтезу гепсидину (рис. 4). Клінічні прояви цих тяжких порушень із перенавантаженням залізом виникають вже у старшому юнацькому віці (ювенільний гемохроматоз) та додатково, окрім печінки, залучають серце (кардіоміопатія). Відкладення заліза в В-клітинах підшлункової залози має деструктивний характер, часто є незворотним і відповідає за виникнення інсулін-залежного діабету. На додачу до цього гіперінсулінемічний діабет може розвинутися через порушений кліренс інсуліну пошкодженими гепатоцитами в пізній стадії хвороби. Однак ситуацію можна поліпшити шляхом проведення флеботомії.
Відкладення заліза в гонадотропних клітинах гіпофізу призводить до гіпогонадотропного гіпогонадизму. Ця не дуже поширена клінічна ознака є більш типовою для осіб із HFE-неасоційованим гемохроматозом. Незалежно від мутації, що лежить в основі, деструктивна артропатія є типовою ознакою при HFE-асоційованому та неасоційованому гемохроматозі і може навіть виникати на фоні лікувальної флеботомії. Патогенетична природа цієї артропатії, що залучає суглоби кисті та опорні суглоби, залишається незрозумілою. Були думки, що вона могла бути викликаною залізо-індукованим пригніченням пірофосфорилаз в синовіальних клітинах, призводячи до відкладення кристалів пірофосфату в суглобових порожнинах.
Література
1. Sharma N., Butterworth J., Cooper B.T. et al. The emerging role of the liver in iron metabolism. Am J Gastroenterol 2005; 100: 201-206.
2. Lam-Yuk-Tseung S., Gros P. Distinct targeting and recycling properties of two isoforms of the iron transporter DMT1 (NRAMP2, Slc11A2). Biochemistry 2006; 45: 2294-2301.
3. Pietrangelo A. Hereditary hemochromatosis – a new look at an old disease. N Engl J Med 2004; 350: 2383-2397.
4. Mack U., Powell L.W., Halliday J.W. A receptor for ferritin on hepatocytes. J Biol Chem 1983; 258: 4672-4675.
5. Hentze M.W., Caughman S.W., Casey J.L. et al. A model for the structure and functions of iron-responsive elements. Gene 1988; 72: 201-208.
6. Johnson D.M., Yamaji S., Tennant J. et al. Regulation of divalent metal transporter expression in human intestinal epithelial cells following exposure to non-haem iron. FEBS Lett 2005; 579: 1923-1929.
7. Andrews N.C. Anemia of inflammation. The cytokine-hepcidin link. J Clin Invest 2004; 113: 1251-1253.
8. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102: 783-788.
9. Nemeth E., Rivera S., Gabayan V. et al. IL‑6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271-1276.
10. Nemeth E., Tuttle M.S., Powelson J. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306: 2090-2093.
11. Pietrangelo A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut 2006; 55: 564-568.
12. Babitt J.L., Huang F.W., Wrighting D.M. et al. Bone morphogenetic protein signalling by hemojuvelin regulates hepcidin expression. Nature Genetics 2006; 38: 531-539.
13. Johnson M.B., Enns C.A. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 2004; 104: 4287-4293.
14. Goswami T., Andrews N.C. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem 2006; in press (manuscript C600197200).
15. Calzolari A., Raggi C., Deaglio S. et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Science 2006; in press (online publication, 17 October 2006).
16. Arredondo M., Tapia V., Rojas A. et al. Apical distribution of HFE-beta2-microglobulin is associated with inhibition of apical iron uptake in intestinal epithelial cells. Biometals 2006; 19: 379-388.
17. Schilsky M.L., Fink S. Inherited metabolic liver disease. Curr Opin Gastroenterol 2006; 22: 215-222.
18. Pietrangelo A., Caleffi A., Henrion J. et al. Juvenile hemochromatosis associated with pathogenetic mutations of adult hemochromatosis genes. Gastroenterology 2005; 128: 470-479.
Stremmel W., Karner M., Manzhalii E., Gilles W., Herrmann T., Merle U. Leber und Eisenstoffwechsel – Uberlegungen zur Pathogenese der genetischen Hamochromatose. Z Gastroenterol 2007; 45(1): 71-75.
* Співавтор та перекладач.
СТАТТІ ЗА ТЕМОЮ Гастроентерологія
Метаболічноасоційована жирова хвороба печінки (МАЖХП) є однією з найактуальніших проблем сучасної гепатології та внутрішньої медицини в цілому. Стрімке зростання поширеності ожиріння та цукрового діабету (ЦД) 2 типу в популяції призвело до істотного збільшення кількості хворих на МАЖХП, яка охоплює спектр патологічних станів від неускладненого стеатозу до алкогольної хвороби печінки та цирозу, що розвиваються на тлі надлишкового нагромадження ліпідів у гепатоцитах. ...
Інфекція Helicobacter pylori (H. pylori) офіційно визнана інфекційним захворюванням і включена до Міжнародної класифікації хвороб (МКХ) 11-го перегляду, тому рекомендовано лікувати всіх інфікованих пацієнтів. Проте, зважаючи на широкий спектр клінічних проявів, пов’язаних із гастритом, викликаним H. pylori, лишаються специфічні проблеми, які потребують регулярного перегляду для оптимізації лікування. ...
Відтворення майбутнього здорової нації – один з найважливіших сенсів існування теперішнього покоління. День боротьби з ожирінням нагадує нам про поширеність цього проблемного явища і важливість попередження його наслідків. Ожиріння може мати вплив на різні аспекти здоров'я, включаючи репродуктивне....
Вивчення клініко-патогенетичних особливостей поєднаного перебігу остеоартрозу (ОА) у хворих із метаболічними розладами, які характеризують перебіг метаболічного синдрому (МС), зокрема цукровим діабетом (ЦД) 2 типу, ожирінням (ОЖ), артеріальною гіпертензією (АГ), є актуальним, оскільки це пов’язано з неухильним збільшенням розповсюдженості цього захворювання, недостатньою ефективністю лікування, особливо за коморбідності з іншими захворюваннями, які патогенетично пов’язані з порушеннями метаболічних процесів. ...