



Застосування модернізованої класифікації ВООЗ (2016) для діагностики мієлоїдних новоутворень

Переглянута класифікація ВООЗ 2016 р. демонструє тісну інтеграцію між даними, отриманими за допомогою морфологічних методів вивчення клітин крові і кісткового мозку і завдяки досягненням молекулярної генетики. Класифікація включає такі нозологічні форми захворювань: 8 типів мієлопроліферативних новоутворень (МПН), для діагностики яких застосовуються нові молекулярно-генетичні критерії; 5 типів мієлодиспластичних/мієлопроліферативних новоутворень (МДН/МПН), у тому числі новий підтип – МДН/МПН з кільцевими сидеробластами і тромбоцитозом (МДН/МПН-КС-Т); 7 форм мієлодиспластичних синдромів (МДС) із новими найменуваннями; 25 підтипів гострих мієлоїдних лейкозів (ГМЛ); новоутворення із бластних плазмоцитоїдних дендритних клітин; гострі лейкози невизначеного походження; В- і Т-лімфобластні лейкози/лімфоми.
Розглянемо основні діагностичні підходи до ідентифікації мієлоїдних новоутворень і гострих лейкозів із застосуванням останньої класифікації ВООЗ.
До групи мієлопроліферативних новоутворень належить низка клональних патологічних процесів, що виникають у результаті трансформації гемопоетичної стовбурової клітини (ГСК) і проліферуючих клітин однієї або більше ліній мієлопоезу (гранулоцитів, мегакаріоцитів, еритроїдних і тучних клітин).
Різні форми МПН (табл. 1) мають низку подібних морфологічних і клініко-гематологічних ознак. Водночас між ними існують значні відмінності, що стосуються клінічних особливостей і даних лабораторного дослідження, важливі в прогностичному плані.

Хронічний мієлолейкоз – одна з найпоширеніших форм МПН. На долю ХМЛ, частота якого складає 1-2 на 100 000 населення щорічно, припадає близько 5-20% усіх лейкозів. ХМЛ діагностується в будь-якому віці, у тому числі і в дітей, але пік захворюваності спостерігається на 5-6‑му десятилітті життя; чоловіки хворіють дещо частіше, ніж жінки.
Практично у всіх хворих на ХМЛ при встановленні діагнозу визначається характерна цитогенетична аномалія – t(9;22)(q34; q11), що призводить до утворення філадельфійської хромосоми (Ph-хромосоми). У 1973 р. Rowley встановила, що Ph-хромосома утворюється в результаті реципрокної транслокації між хромосомами 9 і 22. При цьому протоонкоген ABL, локалізований на довгому плечі хромосоми 9(q34), переноситься на довге плече хромосоми 22 до розташованого в ділянці розриву цієї хромосоми гена BCR. Результатом злиття екзона 2 гена ABL з тією частиною гена BCR, що залишилася на хромосомі 22, є утворення на цій хромосомі химерного гена BCR-ABL.
ХМЛ проходить три фази розвитку: хронічну, акселерації і бластної кризи. У більшості хворих ХМЛ діагностують у хронічній фазі. Її початок не завжди легко визначити, а тривалість може становити 36 міс.
При дослідженні периферичної крові в хронічній фазі ХМЛ спостерігається нейтрофільний лейкоцитоз (12-100×109/л). Важливою гематологічною ознакою за наявності зсуву вліво в лейкограмі є збільшення до 3-4% вмісту базофілів, нерідко при одночасному підвищенні кількості еозинофілів (так звана базофільно-еозинофільна асоціація). При підрахунку лейкограми паличкоядерні і сегментоядерні нейтрофіли складають 35-70%, вміст метамієлоцитів і мієлоцитів (останніх, як правило, більше) коливається між 5 і 40%, промієлоцитів – від 10 до 15%; кількість бластів не перевищує 1-2%.
Для верифікації фази акселерації, або підгострої фази ХМЛ (табл. 2), застосовуються такі лабораторні критерії: вміст мієлобластів у периферичній крові або в кістковому мозку в межах 10-19%; кількість базофілів у периферичній крові ≥20%; не пов’язана з терапією стійка тромбоцитопенія (<100×109/л) або гіпертромбоцитоз (>1000×109/л), що утримується, незважаючи на проведену терапію; нечутливе до терапії тривале підвищення кількості лейкоцитів у крові (>10×109/л) і збільшення розмірів селезінки. Додатковими до гематологічних, морфологічних і цитогенетичних ознак клональної еволюції є низка молекулярно-генетичних параметрів.

Для фази акселерації властиві виражені ознаки дисгранулоцитопоезу, поява в мазках із пунктатів кісткового мозку гіпергранулярних промієлоцитів і мієлоцитів. Спостерігаються й інші прояви дисмієлопоезу, включаючи виявлення кільцевих сидеробластів, набутої пельгерівської аномалії нейтрофілів або еозинофілів, мегакаріоцитів з малими округлими ядрами.
У деяких хворих на ХМЛ у середньому через 4 роки відбувається перехід ХМЛ у гостру фазу з розвитком бластної кризи (БК). Вирішальним для діагностики БК ХМЛ є виявлення ≥20% бластів у периферичній крові й кістковому мозку або наявність екстрамедулярних вогнищ лейкемічної інфільтрації, що складаються виключно з бластних клітин і виявляються в шкірі, лімфатичних вузлах, центральній нервовій системі й інших тканинах і органах.
При БК ХМЛ у 70% хворих субстратні клітини мають мієлоїдну природу, представлені трансформованими клітинами-попередниками гранулоцитарного або еритробластичного й мегакаріоцитарного рядів, або клітинами цих паростків мієлопоезу в різному поєднанні. Приблизно у 20-30% пацієнтів бластні клітини, що виявляються в периферичній крові й кістковому мозку, мають лімфоїдну природу.
Застосування цитохімічних методів дозволяє досить точно визначити природу лейкемічних клітин при БК ХМЛ. При мієлоїдному варіанті БК у бластах спостерігається позитивна реакція при виявленні активності мієлопероксидази (МПО) і нафтол-AS-D-хлорацетатестерази (ХАЕ), слабке дифузне пофарбування цитоплазми клітин при визначенні активності кислої фосфатази (КФ) і негативна реакція при виявленні активності кислої неспецифічної естерази (КНЕ). Для підтвердження мієлоїдної природи бластів проводиться імуноцитохімічне дослідження з використанням моноклональних антитіл (мкАТ) до МПО, антигенів CD13, CD14, CD15, CD33. Взаємодія з мкАТ до антигенів CD41, CD61 і до глікофорину дозволяє ідентифікувати відповідно бластні клітини, що мають ознаки клітин мегакаріоцитарного й еритробластичного рядів.
При лімфоїдному варіанті БК ХМЛ у бластних клітинах не визначається МПО, а при PAS-реакції в цитоплазмі клітин виявляється глікоген у вигляді великих гранул. Лейкемічні клітини при цьому варіанті БК ХМЛ представлені трансформованими В-клітинами-попередниками. На поверхневих мембранах бластних клітин виявляється експресія антигенів CD10, CD19 і CD20.
Хронічний нейтрофільний лейкоз – рідкісне захворювання, що належить до групи МПН, причини виникнення якого залишаються нез’ясованими. Характеризується наявністю анемії, спленомегалії і в низці випадків – гепатомегалії.
При дослідженні периферичної крові визначається виражений нейтрофільний лейкоцитоз (≥25×109/л), більш ніж 80% усіх лейкоцитів крові – сегментоядерні й паличкоядерні нейтрофіли. Вміст незрілих гранулоцитів (промієлоцитів, мієлоцитів, юних) іноді сягає 10%.
У кістковому мозку визначається гіперклітинність, обумовлена проліферацією клітин нейтрофільного ряду, збільшення їх відсоткового вмісту і абсолютної кількості. Лейкоеритроїдне співвідношення становить 20:1 і вище. Вміст бластів і промієлоцитів під час встановлення діагнозу не збільшений. Мієлобласти складають <5% усіх ядровмісних клітин. Можуть спостерігатися також ознаки проліферації клітин еритробластичного й мегакаріоцитарного рядів. При гістологічному вивченні трепанобіоптатів кісткового мозку дуже рідко виявляються ознаки фіброзу.
При цитохімічному дослідженні в лейкоцитах при ХНЛ, на відміну від ХМЛ, визначається підвищена активність лужної фосфатази. У кровотворних клітинах хворих ХНЛ не визначається Ph-хромосома або злитий ген BCR/ABL і перебудови генів PDGFRA, PDGFRB, FGFR1, або PCM1-JAK2. Виявляються асоційовані з цією формою МПН мутації гена CSF3R або інші мутації, пов’язані з активацією CSF3R (табл. 3). За відсутності мутацій гена CSF3R діагноз ХНЛ може ґрунтуватися на виявленні стійкої нейтрофілії (упродовж принаймні 3 міс).

Диференціальна діагностика ХНЛ після виключення інфекційних і запальних процесів, що супроводжуються нейтрофільним лейкоцитозом, проводиться з іншими формами МПН – справжньою поліцитемією, первинним мієлофіброзом, есенціальною тромбоцитемією. Відсутність ознак дисплазії в клітинах гранулоцитарного ряду і мієлодиспластичних змін у клітинних елементах інших ліній мієлопоезу, а також вміст моноцитів у крові <1×109/л дозволяє відрізнити ХНЛ від різних форм МДС, хронічного мієломоноцитарного лейкозу (ХММЛ), інших МДН/МПН.
Справжня поліцитемія (еритремія, синдром Вакеза-Ослера) – рідкісне хронічне МПН, для якого характерне надмірне утворення еритроцитів незалежно від механізмів, що регулюють еритропоез у нормі.
Частота захворюваності становить 0,5-1 на 100 тис. населення в рік. Співвідношення чоловіків і жінок – 1,2:1, а середній вік хворих – 60-70 років.
Етіологія захворювання досі залишається нез’ясованою. Значення генетичної схильності підтверджують випадки сімейної СП. У розвитку захворювання в окремих категорій хворих не виключена роль іонізуючої радіації, токсичних чинників і вірусів.
Практично всі хворі на СП є носіями мутацій V617F в гені янус-кінази 2 (JAK2) та іншої функціонально подібної мутації JAK2, що призводить до проліферації не лише клітин еритробластичного ряду, але й гранулоцитів і мегакаріоцитів. Вважають, що в основі виникнення СП лежить трансформація гемопоетичної стовбурової клітини.
У розвитку СП виділяють три послідовні стадії: продромальну, передполіцитемічну фазу, основною ознакою якої є збільшення маси еритроцитів; фазу стабільного перебігу захворювання, що асоціюється зі значним збільшенням маси еритроцитів; стадію постполіцитемічного мієлофіброзу, при якій розвиток цитопенії, включаючи анемію, обумовлений неефективним гемопоезом, гіперспленізмом і появою вогнищ екстрамедулярного кровотворення.
У 90% хворих відзначається еритроціанотичне забарвлення шкіри і слизових оболонок, часті тромбози судин (майже у 30% хворих) і геморагічні прояви (у 25% пацієнтів). До числа основних за значущістю й частотою клінічних ознак належить спленомегалія (у 80% випадків) і гепатомегалія (у 70% хворих).
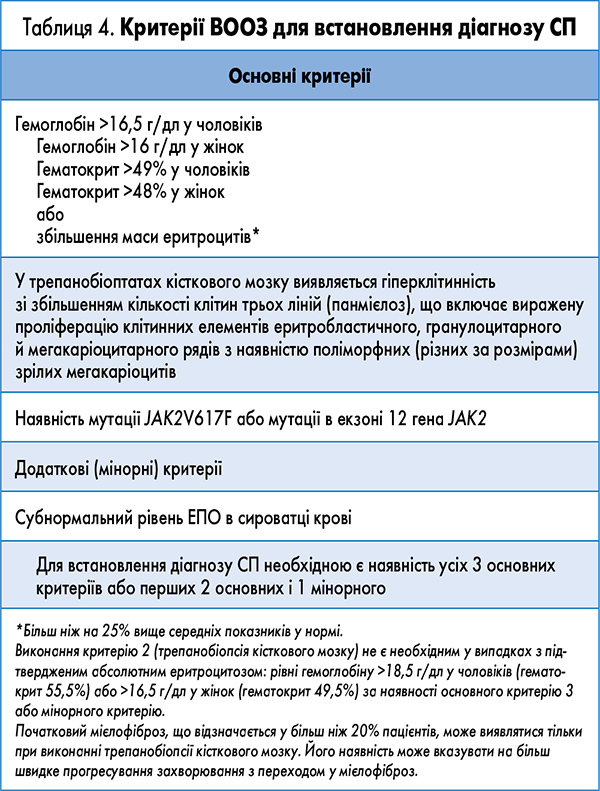
Основним з клініко-лабораторних досліджень для встановлення діагнозу СП є збільшення кількості еритроцитів (6-7×1012/л) і рівня гемоглобіну (180-220 г/л), що супроводжуються зростанням показників гематокриту >49% у чоловіків і >48% у жінок.
У мазках зі стернального пунктату кісткового мозку визначається гіперплазія, обумовлена збільшенням кількості клітинних елементів не лише нормобластичного еритропоезу, але й клітин гранулоцитарного ряду. Визначається також проліферація поліморфних зрілих мегакаріоцитів, різних за величиною.
СП діагностують, керуючись такими критеріями (А, В), запропонованими експертами ВООЗ:
А1. Збільшення маси циркулюючих еритроцитів >25% (≥36 мл/кг у чоловіків, 32 мл/кг у жінок) або вмісту гемоглобіну (>185 г/л у чоловіків, 165 г/л у жінок).
А2. Мутації гена JAK2V617F або в екзоні 12 активованого JAK2.
А3. Виключення причин, що викликають вторинний еритроцитоз, у тому числі: відсутність сімейного еритроцитозу; відсутність підвищеного рівня еритропоетину (ЕПО), викликаного гіпоксією – насичення артеріальної крові киснем (РО2) ≤92%, гемоглобіном з підвищеною спорідненістю до кисню, неефективністю рецептора ЕПО, невідповідним виробленням ЕПО пухлиною.
А4. Спленомегалія.
А5. Утворення ендогенних еритроїдних колоній in vitro.
В1. Тромбоцитоз >400×109/л.
В2. Кількість лейкоцитів >12×109/л.
В3. При трепанобіопсії кісткового мозку виявляється панмієлоз з вираженою проліферацією клітин еритробластичного й мегакаріоцитарного рядів.
В4. Субнормальний рівень ЕПО в сироватці крові.
Діагноз СП встановлюється за наявності А1+А2 і будь-якої іншої ознаки з категорії А або А1+А2 і будь-яких двох ознак з категорії В.
У 10-20% хворих спостерігається перехід у фазу постполіцитемічного мієлофіброзу, для якої характерне зменшення маси клітин червоної крові, що виявляється при радіоізотопному дослідженні, збільшення міри спленомегалії, посилення фіброзу кісткового мозку зі збільшенням кількості ретикулінових і колагенових волокон, розвиток цитопенії. Можуть спостерігатися диспластичні зміни в клітинах еритробластичного і гранулоцитарного рядів, що не виявлялися раніше. Вміст бластів у крові й кістковому мозку, як правило, не досягає 10%. Розвиток вогнищ екстрамедулярного гемопоезу поєднується зі збільшенням розмірів селезінки.
Диференціальна діагностика СП проводиться за наявності низки захворювань і станів, що супроводжуються вторинним (симптоматичним) еритроцитозом. В онкогематологічній практиці особливої важливості набуває диференціальна діагностика СП у стабільній фазі захворювання і вторинних абсолютних еритроцитозів, що зустрічаються у хворих з гіпернефромою, пухлинами нирок, ендокринних органів (табл. 4).
Первинний мієлофіброз (синоніми: хронічний ідіопатичний мієлофіброз, агногенна мієлоїдна метаплазія, ідіопатичний мієлофіброз) – клональне МПН, в основі розвитку якого лежить трансформація стовбурових кровотворних клітин кісткового мозку. Характерні ознаки захворювання – переважна проліферація клітин мегакаріоцитарного і гранулоцитарного рядів у кістковому мозку, що супроводжується розвитком фіброзу й остеосклерозу, спленомегалія, поява вогнищ екстрамедулярного гемопоезу, анемія, зміни в лейкоцитарній формулі крові.
Захворюваність на ПМФ у різних країнах становить 0,5-1,5 на 100 тис. населення. Хворіють переважно особи літнього віку (60-70 років). До числа найважливіших клінічних ознак належать спленомегалія (у 90% хворих) і гепатомегалія (у 50% випадків).
При розвитку ПМФ відзначається еволюція – від початкової передфібротичної стадії захворювання, що характеризується гіперклітинністю кісткового мозку й відсутністю мінімальних ознак ретикулінового фіброзу (табл. 5), до стадії вираженого колагенового фіброзу й остеосклерозу.

При рутинному аналізі периферичної крові визначається нормохромна анемія, лейкоцитоз та/або тромбоцитоз, наявність ядровмісних клітин еритробластичного ряду. Кількість лейкоцитів у 40% хворих коливається в межах 11-25×109/л, у деяких перевищує 40×109/л. У крові хворих на ПМФ зустрічаються гіпер- і гіпосегментовані лейкоцити, невеликий відсоток незрілих клітин гранулоцитарного ряду (мієлоцитів і промієлоцитів). Показники активності лейкоцитарної лужної фосфатази в нейтрофілах, що виявляється при цитохімічному дослідженні, як правило, підвищені, але в низці випадків може спостерігатися її нормальний рівень і навіть зниження ферментативної активності.
У багатьох хворих на ПМФ може визначатися підвищена кількість тромбоцитів (до 500×109/л). У мазках периферичної крові виявляються атипові, великі і гіпогранулярні тромбоцити, аномальні мегакаріоцити й голі ядра мегакаріоцитів.
При аналізі мієлограми визначається дифузна або осередкова гіперклітинність кісткового мозку – представлені клітинні елементи усіх трьох основних ліній мієлопоезу, хоча в окремих ділянках зрізів можуть переважати клітини того або іншого типу. У 90% хворих визначається проліферація атипових мегакаріоцитів, що утворюють кластери з 3-10 клітин (табл. 6). Вона супроводжується наявністю фіброзу зі збільшенням ретикулінових та/або колагенових волокон.
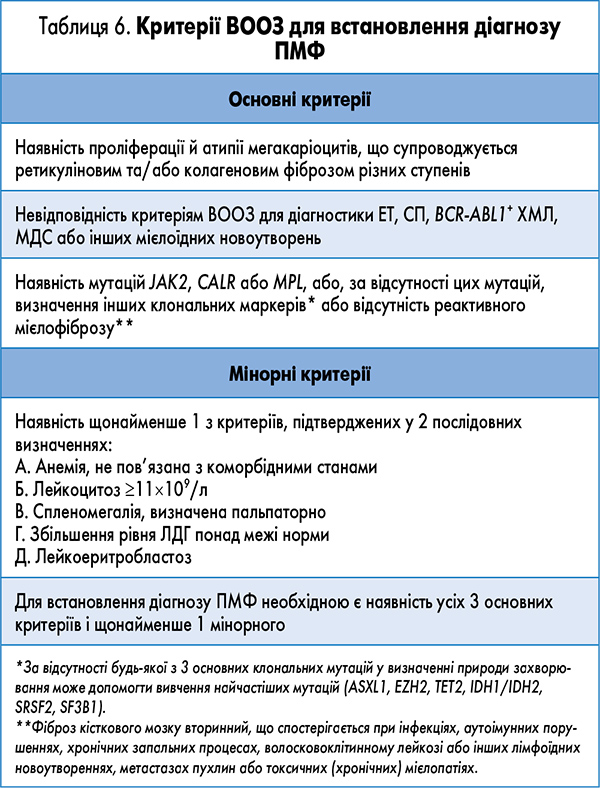
Поява у хворих на ПМФ 10-19% бластів у периферичній крові та/або кістковому мозку і визначення на основі імуноцитохімічного дослідження збільшеної кількості CD34+ клітин, що утворюють кластери поблизу ендосту, вказує на перехід захворювання у фазу акселерації. Наявність 20% і більше бластів свідчить про трансформацію захворювання в гострий лейкоз мієлоїдного походження.
Генетичні дефекти, специфічні тільки для ПМФ, неідентифіковані. Приблизно у 50% хворих на ПМФ виявляються мутації V617F гена JAK2.
Есенціальна тромбоцитемія (синоніми: первинний тромбоцитоз, ідіопатичний тромбоцитоз, геморагічна тромбоцитемія) – клональне мієлопроліферативне новоутворення, що характеризується переважним ураженням клітин мегакаріоцитарного ряду і збільшенням кількості тромбоцитів у периферичній крові (≥450×109/л).
Показники щорічної захворюваності становлять 0,6‑2,5 на 100 тис. населення. Хворіють переважно люди віком 50‑60 років. Можливе джерело виникнення – трансформована гемопоетична стовбурова клітина (ГСК). Етіологія й механізми розвитку захворювання ще недостатньо вивчені.
Майже в половини хворих на ЕТ спостерігається безсимптомний перебіг захворювання. У деяких пацієнтів у клінічній картині на перший план виходять ускладнення, обумовлені тромбозом судин і крововиливами. У 50% хворих визначається спленомегалія, а у 15-20% – ознаки гепатомегалії. Кількість тромбоцитів у периферичній крові хворих збільшена й нерідко перевищує 1000×109/л (табл. 7). Для тромбоцитів характерні ознаки анізоцитозу, нерідко виявляються атипові великі форми, агранулярні тромбоцити. Зрідка в крові зустрічаються ядровмісні фрагменти мегакаріоцитів.

Рівень гемоглобіну у хворих на ЕТ коливається в межах 100-188 г/л (у середньому 138 г/л). Еритроцити нормоцитарні й нормохромні; середня кількість лейкоцитів у периферичній крові становить 11,5×109/л, але можливі коливання – від 6 до 41×109/л, причому незрілі клітини гранулоцитарного ряду в мазках периферичної крові виявляються вкрай рідко.
При дослідженні мазків зі стернального пунктату або гістологічному вивченні трепанобіоптатів кісткового мозку визначається нормоклітинність або гіперклітинність кісткового мозку. Спостерігається виражена проліферація клітин мегакаріоцитарного ряду з переважанням великих або велетенських форм з гіперчасточковими ядрами й широкою цитоплазмою. Мегакаріоцити в мазках і зрізах розташовуються рівномірно або утворюють скупчення у вигляді кластерів. Атипові мегакаріоцити, характерні для ПМФ, при ЕТ не виявляються.
Гістологічне вивчення трепанобіоптатів кісткового мозку є особливо цінним для виключення деяких форм МДС, що супроводжуються збільшенням кількості тромбоцитів у крові, таких як МДС, асоційований з ізольованою del(5q), і МДН/МПН-КС-Т.
Специфічні для ЕТ цитогенетичні аномалії не встановлені. У 40-50% хворих визначаються мутації V617F гена JAK2, що відбуваються на рівні ГСК, або функціонально подібні мутації, що не виявляються при реактивному тромбоцитозі. При діагностиці ЕТ враховується також наявність мутацій генів CALR і MPL.
Перебіг ЕТ індолентний, з тривалими безсимптомними інтервалами з епізодами, що супроводжуються тромбоемболічними або геморагічними проявами. Медіана виживаності хворих становить 10-15 років. Трансформація в ГМЛ або МДС, пов’язана переважно з попередньою цитотоксичною терапією, відбувається менш ніж у 5% хворих.
Хронічний еозинофільний лейкоз (ХЕЛ) – це МПН, за якого автономна клональна проліферація клітин – попередників еозинофільного ряду призводить до перманентного збільшення кількості еозинофілів у периферичній крові, кістковому мозку, інших органах і тканинах. До групи хворих на ХЕЛ НІЧ не належать хворі з Ph-хромосомою, злитним геном BCR-ABL1 або перебудовою генів PDGFRA, PDGFRB і EGFR1 (табл. 8).
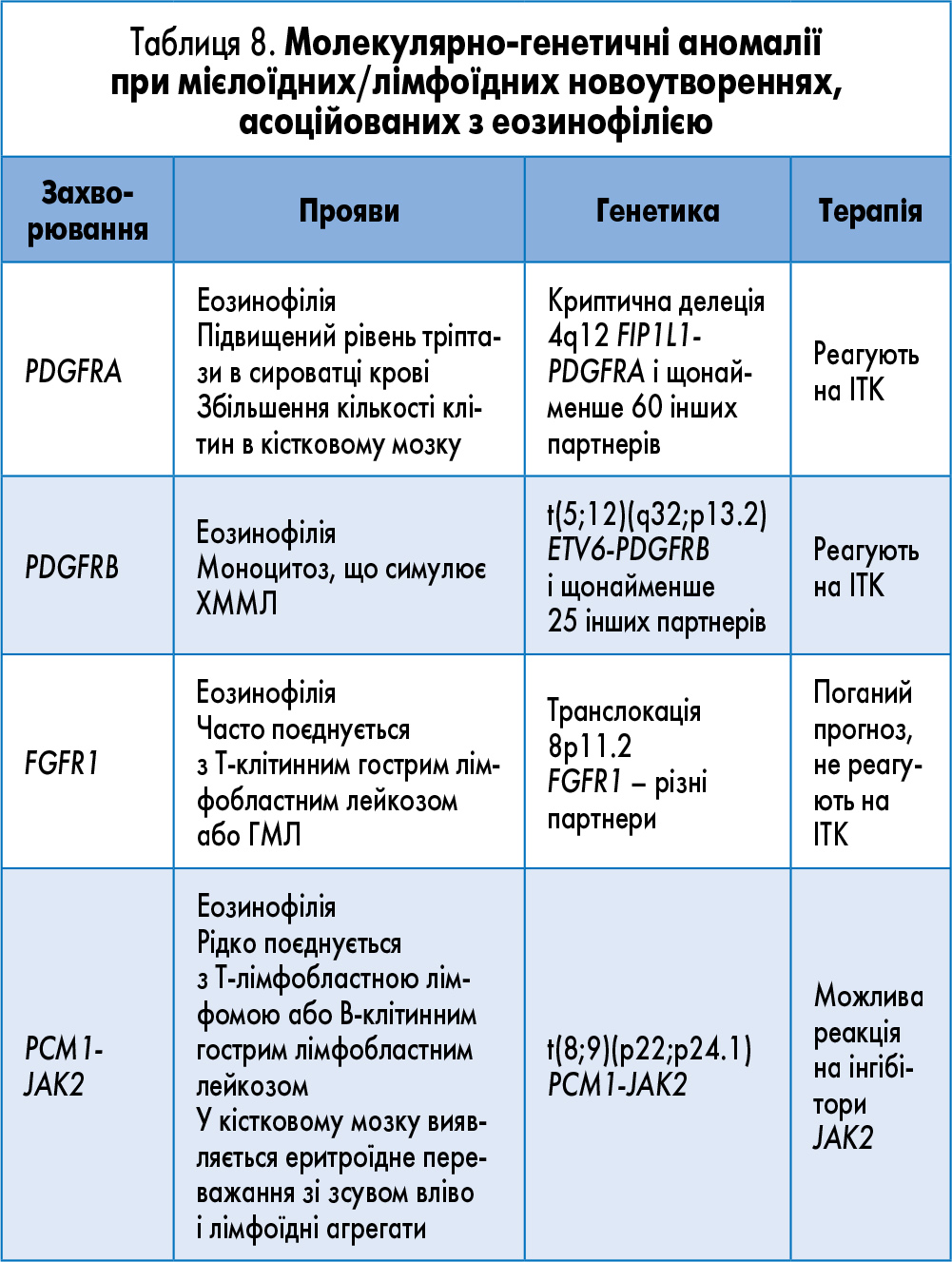
Вміст еозинофілів у крові пацієнтів з ХЕЛ НІЧ, як правило, >1,5×109/л. Кількість бластів у периферичній крові й кістковому мозку <20%.
Для встановлення діагнозу ХЕЛ необхідно довести клональний характер еозинофілії або виявити збільшення кількості мієлобластів у крові й кістковому мозку. У багатьох випадках це неможливо, і тоді перевага надається діагнозу «ідіопатичний гіпереозинофільний синдром». Для його встановлення достатнім є виявлення абсолютної еозинофілії (≥1,5×109/л) упродовж 6 міс спостереження за пацієнтом. При цьому не вдається з’ясувати причину стійкого збільшення кількості еозинофілів у периферичній крові (необхідно виключити патологічні процеси, що супроводжуються реактивною еозинофілією, спричиняються паразитами й іншими інфекційними агентами, алергічні захворювання, колагенози). З-поміж захворювань неопластичної природи мають бути виключені Т-клітинні неходжкінські лімфоми, лімфома Ходжкіна, множинна мієлома, метастази пухлин у різних органах.
Приблизно у 10% пацієнтів з ХЕЛ НІЧ захворювання протікає безсимптомно, і еозинофілія виявляється випадково за даними аналізу крові. У 30-50% випадків можуть спостерігатися ознаки спленомегалії й гепатомегалії. Для частини хворих характерними є анемія та тромбоцитопенія.
У периферичній крові містяться переважно зрілі еозинофіли й невелика кількість еозинофільних мієлоцитів і промієлоцитів. У деяких випадках відзначається гіпер- або гіпосегментація ядер, збільшення їх розмірів, вакуолізація цитоплазми клітин і зменшена кількість гранул у ній. У частини пацієнтів може бути помірний нейтрофільоз або моноцитоз. В окремих випадках виявляється помірна базофілія.
Кістковий мозок є гіперклітинним за рахунок збільшення вмісту еозинофілів, причому представлені всі стадії дозрівання клітин цього ряду. Вміст клітин еритробластичного й мегакаріоцитарного рядів у межах норми. На користь діагнозу ХЕЛ НІЧ свідчить збільшення кількості мієлобластів (5-19%) і наявність диспластичних змін у клітинах інших ліній.
Виявлення клональних хромосомних аномалій, таких як трисомія 8 та i(17q), у складних випадках дозволяє диференціювати ХЕЛ та ідіопатичний еозинофільний синдром (ІЕС). У низці випадків з ХЕЛ асоціюються такі цитогенетичні аномалії, як t(8;13)(p11; q12) та інші транслокації 8p11 – t(8;9)(p11; q32-34) і t(6;8)(q27; p11). У деяких хворих виявляються мутації гена JAK2.
До категорії мієлодиспластичних/мієлопроліферативних новоутворень належать клональні процеси, які до моменту маніфестації мають клінічні, лабораторні й морфологічні ознаки, що перекриваються і характерні як для МДН, так і для МПН.
До групи МДН/МПН, відповідно до модифікованої у 2016 р. класифікації ВООЗ, належать ХММЛ, атиповий BCR-ABL1 негативний хронічний мієлолейкоз (аХМЛ), ювенільний мієломоноцитарний лейкоз (ЮММЛ), мієлодиспластичне/мієлопроліферативне новоутворення некласифіковане (МДН/МПН-Н) і як «попередня» нозологічна форма – МДН/МПН з кільцевими сидеробластами і тромбоцитозом (МДН/МПН-КС-Т), раніше відома як рефрактерна анемія з кільцевими сидеробластами і тромбоцитозом (РАКС-Т). Критеріями для діагностики МДН/МПН-КС-Т визнані: наявність рефрактерної анемії, ознак дизеритропоезу в кістковому мозку, вміст кільцевих сидеробластів, що становлять ≥15% серед ядровмісних клітин еритробластичного ряду, виявлення мегакаріоцитів з ознаками, що спостерігаються при первинному мієлофіброзі й есенціальній тромбоцитемії, кількість тромбоцитів у периферичній крові >450×109/л.
Після виявлення зв’язку МДН/МПН-КС-Т з мутаціями гена SF3B1 (які своєю чергою асоційовані з наявністю кільцевих сидеробластів) з’явилися нові докази на підтримку того, що МДН/МПН-КС-Т може бути самостійною нозологічною формою.
У хворих на МДН/МПН-КС-Т мутації гена SF3B1 часто поєднуються з мутаціями JAK2V617F і менш часто (<10%) з мутаціями генів CALR або MPL.
Хронічний мієломоноцитарний лейкоз характеризується персистентним моноцитозом – >10% у лейкограмі при абсолютній кількості моноцитів у периферичній крові >1×109/л, відсутністю злитого гена BCR-ABL і перебудов генів PDGFRA, PDGFRB, FCFR1. Кількість бластів і промоноцитів у крові і кістковому мозку не досягає 20%. У клітинах одного або більше паростків мієлопоезу відзначаються диспластичні зміни. У хворих, як правило, спостерігається збільшення кількості лейкоцитів і абсолютний моноцитоз (моноцити є зрілими, з незміненими цитоморфологічними ознаками). Промієлоцити й мієлоцити складають <10% від загальної кількості лейкоцитів. У більшості хворих виявляються ознаки дисгранулоцитопоезу, що виражаються в появі нейтрофілів з гіпо- і аномально часточковими ядрами, наявність аномальної зернистості в цитоплазмі (табл. 9).

У модифікованій класифікації ВООЗ (2016) пропонується поділяти ХММЛ на 3 групи залежно від відсотка бластів і промоноцитів у кістковому мозку й периферичній крові.
До категорії ХММЛ‑0 належать випадки з наявністю <2% бластів у периферичній крові і <5% – у кістковому мозку.
При ХММЛ‑1 вміст бластних клітин у крові становить 2-4% та/або 5-9% – у кістковому мозку.
При ХММЛ‑2 кількість бластів у крові становить 5-19% і в кістковому мозку – 10-19% та/або в цитоплазмі наявні клітини паличок Ауера.
У зв’язку із встановленням клінічних і молекулярно-генетичних відмінностей між так званими проліферативним (кількість лейкоцитів ≥13×109/л) і диспластичним типами ХММЛ (кількість лейкоцитів <13×109/л), і особливо відмінностей, пов’язаних з аберацією в RAS/MPK сигнальних шляхах, виправданим є виділення цих двох підтипів захворювання.
У мазках зі стернального пунктату кісткового мозку більш ніж у 75% пацієнтів визначається гіперклітинність. У більшості хворих при дослідженні кісткового мозку виявляються ознаки дисгранулоцитопоезу, а майже у 50% – дизеритропоезу (часточковість ядер, поява клітин з мегалобластоїдними ознаками). Ознаки дисмегакаріоцитопоезу спостерігаються у 70-80% пацієнтів і проявляються в наявності мікромегакаріоцитів або мегакаріоцитів з аномальною часточковістю ядер. Майже у третини хворих при гістологічному вивченні трепанобіоптатів кісткового мозку виявляються ознаки фіброзу.
При ХММЛ найчастіше спостерігаються мутації генів SRSF2, TET2 та/або ASXL1 (>80% випадків). Наявність мутацій гена ASXL1 обумовлює більш агресивний перебіг захворювання.
Атиповий хронічний мієлолейкоз, BCR-ABL1 негативний – захворювання, якому на момент встановлення діагнозу властиві як мієлопроліферативні, так і мієлодиспластичні ознаки. Наявність мультилінійних ознак дисплазії вказує на те, що в основі розвитку захворювання лежить ураження мієлоїдної стовбурової клітини кісткового мозку. Статистичні дані щодо частоти аХМЛ практично відсутні. Вважають, що на кожні 100 випадків Ph-позитивного ХМЛ припадає 1-2 хворих на аХМЛ.
У більшості хворих спостерігаються ознаки сплено- й гепатомегалії. Основні прояви обумовлені наявністю анемії, в окремих випадках – тромбоцитопенії. Кількість лейкоцитів у периферичній крові, як правило, підвищена, коливається в межах 24-69×109/л, у деяких випадках >300×109/л (табл. 10). Вміст бластів у периферичній крові у більшості хворих <5% і ніколи не досягає 20%. Кількість незрілих гранулоцитів (промієлоцитів, мієлоцитів, метамієлоцитів) у лейкограмі становить 10-20%, іноді дещо вища. Абсолютна кількість моноцитів у лейкограмі рідко перевищує 10%. Кількість базофілів трохи підвищена.

Характерною рисою аХМЛ є дисгранулоцитопоез, що проявляється в наявності аномальної зернистості в цитоплазмі нейтрофілів та псевдопельгерівських лейкоцитів.
У гістологічних зрізах трепанобіоптатів кісткового мозку визначається гіперклітинність, обумовлена підвищеним вмістом зрілих і незрілих нейтрофілів. Помірно збільшена кількість бластів, але їх вміст завжди <20%. Кількість мегакаріоцитів може бути зниженою або збільшеною. У деяких з них спостерігаються диспластичні зміни. В окремих випадках клітинні елементи еритропоезу, у тому числі і з дизеритропоетичними ознаками, можуть становити до 50%. Збільшена кількість ретикулінових волокон може виявлятися в момент встановлення діагнозу або пізніше при прогресії захворювання.
За даними цитохімічного дослідження, активність лужної фосфатази, різко знижена в нейтрофілах при Ph‑позитивному ХМЛ, у хворих на аХМЛ може бути низькою, не відрізнятися від показників у нормі і навіть бути підвищеною. При реакції на α-нафтілацетатестеразу й кислу неспецифічну естеразу в кістковому мозку хворих на аХМЛ виявляється більший вміст клітин моноцитарного ряду, ніж при забарвленні за Папенгеймом.
Каріотипічні аномалії виявляються у 80% хворих на аХМЛ. Найчастішими є такі аномалії, як +8 і del(20q), але зустрічаються також аномалії хромосом 13, 14, 17, 19 і 12. Зрідка в пацієнтів з аХМЛ визначається i(17q), більш властивий для ХММЛ. Майже у 30% хворих виявляються набуті мутації генів NRAS і KRAS, у деяких випадках – мутації JAK2V617F.
Підтип аХМЛ нині досить повно охарактеризований на молекулярному рівні, що дозволяє легше відрізнити його від ХНЛ. Якщо ХНЛ значною мірою асоційований з мутаціями гена CSF3R, то при аХМЛ вони зустрічаються вкрай рідко (<10%). І навпаки, аХМЛ асоціюється з мутаціями генів SETBP1 та/або ETNK1 майже у третини пацієнтів.
Ювенільний мієломоноцитарний лейкоз – агресивне клональне неопластичне захворювання кровотворної тканини, що зустрічається в немовлят або в дитячому віці й характеризується переважною проліферацією клітин гранулоцитарного і моноцитарного рядів (табл. 11). Аномалії клітин еритробластичного й мегакаріоцитарного рядів, що часто виявляються при цьому, слугують непрямим підтвердженням того, що в основі розвитку лейкемічного процесу лежить трансформація мієлоїдної стовбурової клітини кісткового мозку.

Захворюваність на ЮММЛ – приблизно 0,13 на 100 тис. дитячого населення. ЮММЛ становить 2-3% усіх випадків лейкозів у дітей або 20-30% усіх випадків мієлодиспластичних і мієлопроліферативних захворювань у пацієнтів до 14 років. Основними клінічними проявами захворювання є анемія, спленомегалія, гепатомегалія (75%), лімфаденопатія (у 20% хворих), шкірні прояви у вигляді висипання, обумовлені лейкемічними інфільтратами, специфічні прояви депігментації шкіри. У частини хворих відзначається гарячкова реакція, симптоми тонзиліту або бронхіту, ознаки геморагічного діатезу.
Клінічні прояви у хворих на ЮММЛ часто нагадують ті, що спостерігаються при інфекційних захворюваннях, викликаних вірусом Епштейна-Барр, цитомегаловірусом, вірусом герпесу людини 6 типу тощо. Уже на початку захворювання значно зменшується кількість тромбоцитів (до 40×109/л). Загальна кількість лейкоцитів у крові, як правило, збільшена, але нижче, ніж при Ph+ ХМЛ і коливається в межах 25-35×109/л. Лише у 5-10% хворих вміст лейкоцитів у крові >100×109/л. Лейкоцитоз обумовлений збільшенням кількості нейтрофільних лейкоцитів і моноцитів.
У лейкограмі виявляються незрілі форми гранулоцитів, у тому числі мієлоцити і промієлоцити. Може визначатися підвищена кількість базофілів. У мазках периферичної крові можуть виявлятися незрілі клітини еритробластичного ряду, кількість яких збільшується при прогресії захворювання. Зустрічаються окремі плазматичні клітини й імунобласти. Сумарний вміст бластних клітин і промоноцитів у периферичній крові завжди <20%.
Кістковий мозок є гіперклітинним, містить збільшену кількість бластів (до 10-15%), незрілих і зрілих гранулоцитів. Моноцити становлять 5-10% від загальної кількості мієлокаріоцитів, але в окремих хворих їх вміст може бути підвищений до 30%. Вміст бластів і промоноцитів <20%. У деяких випадках у клітинах гранулоцитарного й еритробластичного рядів можуть виявлятися нерізко виражені диспластичні зміни (псевдопельгерівські лейкоцити, гіпогрануляція цитоплазми нейтрофілів, мегалобластоїдні зміни в клітинах еритробластичного ряду). Вміст мегакаріоцитів, як правило, знижений, ознаки дисплазії в клітинах мегакаріоцитарного ряду спостерігаються рідко.
Вогнища лейкемічної інфільтрації, представлені трансформованими клітинами гранулоцитарного й моноцитарного рядів, виявляються також у поверхневих і глибоких шарах шкіри і в легенях.
Майже у 90% хворих спостерігаються мутації генів PTPN11, KRAS, NRAS, CBL або NF1. При аналізі каріотипу майже у 20% хворих на ЮММЛ виявляється моносомія 7 хромосоми, у 10% пацієнтів – інші аномалії.
У цілому діагностичні критерії ЮММЛ включають клінічні й гематологічні ознаки, дані цитогенетичних і молекулярно-генетичних досліджень і деякі додаткові умови. До перших належать: кількість моноцитів у периферичній крові ≥1×109/л, вміст бластів у крові і кістковому мозку <20%, наявність спленомегалії, відсутність Ph-хромосоми (перебудов BCR-ABL1). Щодо генетичних аномалій, достатньо наявності однієї з таких: мутації генів PTPN11, або KRAS, або NRAS.
Критерії третьої групи повинні використовуватися при діагностиці ЮММЛ у хворих за відсутності молекулярно-генетичних змін: наявність моносомії 7 або інших аномалій хромосом і принаймні двох таких: вміст гемоглобіну F, що збільшується з віком, визначення в мазках крові мієлоїдних і еритроїдних клітин-попередників, гіперчутливість до гранулоцитарно-макрофагального колонієстимулювального фактора при дослідженні колоній, гіперфосфорилювання STAT5.
МДН/МПН з кільцевими сидеробластами і тромбоцитозом. Раніше ця форма була відома як РАКС-Т. Характеризується рефрактерною анемією, дизеритропоезом у кістковому мозку з наявністю кільцевих сидеробластів і мегакаріоцитів з ознаками, що спостерігаються при первинному мієлофіброзі й есенціальній тромбоцитемії. Основою для виділення МДН/МПН-КС-Т як самостійної нозологічної форми слугує часта асоціація з мутаціями гена SF3B1 (з якими, у свою чергу, пов’язана наявність кільцевих сидеробластів). При МДН/МПН-КС-Т мутації SF3B1 часто поєднуються з мутаціями V617F генів JAK2 і рідше (<10%) – з мутаціями CALR і MPL (табл. 12).

Мієлодиспластичне/мієлопроліферативне новоутворення некласифіковане. Діагноз МДН/МПН-Н встановлюється у хворих за наявності клініко-лабораторних і морфологічних ознак, властивих як для МДС, так і для мієлопроліферативних захворювань, але за відсутності критеріїв, що дозволяють віднести їх до зазначених вище форм МДН/МПН (ХММЛ, аХМЛ, ЮММЛ). Характеризується неефективною проліферацією диспластичних клітин однієї з ліній мієлопоезу. Діагностування МДН/МПН-Н має важливе клінічне значення, бо визначає вибір терапії.
При захворюванні вражаються селезінка, печінка, є вогнища лейкемічної інфільтрації в інших органах. При дослідженні крові визначається анемія з ознаками макроцитозу або без них. Результати лабораторних досліджень при встановленні діагнозу МДН/МПН-Н мають бути такими.
• Наявність клінічних, лабораторних і морфологічних ознак, властивих одній із форм МДН (МДН з ознаками однолінійної дисплазії, МДН з кільцевими сидеробластами, МДН з мультилінійною дисплазією, МДН з надлишком бластів), <20% бластів у крові й кістковому мозку.
• Виражені ознаки мієлопроліферативного процесу: кількість тромбоцитів ≥450×109/л поєднується з проліферацією мегакаріоцитів, або кількість лейкоцитів ≥13×109/л; наявність або відсутність вираженої спленомегалії.
• Відсутність в анамнезі хворого вказівок на раніше діагностоване МПН або МДН.
• Відсутність даних щодо проведеної терапії цитотоксичними препаратами або ростовими факторами, які могли б пояснити появу мієлодиспластичних або мієлопроліферативних ознак. Відсутність Ph-хромосоми або злитного гена BCR-ABL1, t(3;3)(q21; q26), del(5q) або inv(3)(q21; q26).
• Наявність захворювання, що виникло de novo, зі змішаними мієлопроліферативними і мієлодиспластичними ознаками, що не дають можливості віднести його до тієї або іншої категорії МДН, МПН або МДН/МПН.
СТАТТІ ЗА ТЕМОЮ Онкологія та гематологія
Гостра лімфобластна лейкемія (ГЛЛ) є найпоширенішим онкогематологічним захворюванням у дітей і складає значну частку серед лейкемій у дорослих. Незважаючи на значні успіхи в лікуванні ГЛЛ у дітей, де рівень виліковності сягає 90%, результати терапії у дорослих залишаються незадовільними. У рамках науково-практичної конференції з міжнародною участю «Діагностика та лікування гематологічних захворювань: підведення підсумків 2023 року» (15-16 грудня 2023 року) проведено секцію, присвячену ГЛЛ....
Хронічна лімфоцитарна лейкемія (ХЛЛ) залишається актуальною проблемою сучасної онкогематології. Незважаючи на певні досягнення в терапії, ХЛЛ є невиліковним захворюванням. Стандартна хіміотерапія не забезпечує стійкої відповіді, а трансплантація гемопоетичних стовбурових клітин можлива лише для окремої когорти пацієнтів. Тому пошук нових підходів до терапії ХЛЛ, зокрема таргетної, є нагальним завданням. ...
Гепатоцелюлярна карцинома (ГЦК) – злоякісне новоутворення в печінці, що розвивається з гепатоцитів. Рання діагностика і початок лікування пацієнтів із ГЦК запобігає виникненню тяжких ускладнень і покращує якість життя пацієнтів. Медична допомога пацієнтам із ГЦК потребує міждисциплінарної співпраці та інтегрованого ведення хворих мультидисциплінарною командою фахівців, яка займається або спеціалізується на злоякісних новоутвореннях печінки. Саме цьому сприятимуть положення Стандарту медичної допомоги «Гепатоцелюлярна карцинома»....
Традиційно січень є місяцем обізнаності про рак шийки матки (РШМ) – однієї з найпоширеніших патологій у структурі онкогінекологічних захворювань. Протягом цього місяця світ забарвлюється в палітру бірюзового та білого з метою привернення уваги громадськості до проблеми РШМ. ...