


6 квітня, 2018
Клінічні маски лейкодистрофій у дитячому віці
Хвороба Канаван та метахроматична лейкодистрофія
 Лейкодистрофії становлять групу нейродегенеративних захворювань, які уражують переважно білу речовину головного мозку та можуть викликати тяжкі неврологічні розлади у дітей. Важливим методом для підтвердження діагнозу є магнітно-резонансна томографія (МРТ) головного мозку, проте, незважаючи на існування сучасних методів нейровізуалізації, своєчасна діагностика цих захворювань залишається складною проблемою. Лейкодистрофії часто перебігають під клінічними масками інших неврологічних і психіатричних розладів.
Лейкодистрофії становлять групу нейродегенеративних захворювань, які уражують переважно білу речовину головного мозку та можуть викликати тяжкі неврологічні розлади у дітей. Важливим методом для підтвердження діагнозу є магнітно-резонансна томографія (МРТ) головного мозку, проте, незважаючи на існування сучасних методів нейровізуалізації, своєчасна діагностика цих захворювань залишається складною проблемою. Лейкодистрофії часто перебігають під клінічними масками інших неврологічних і психіатричних розладів.
Від початку клінічних проявів до постановки правильного діагнозу пацієнти зазвичай проходять довгий шлях. Для своєчасного розпізнавання цих станів необхідне знання клінічних особливостей їх перебігу, а також можливостей інструментальної, лабораторної та генетичної діагностики, результати яких можуть бути вирішальними у встановленні правильного діагнозу. Ми наводимо приклади з власного досвіду щодо діагностичного шляху постановки діагнозу в дітей із двома формами лейкодистрофій – хворобою Канаван та метахроматичною лейкодистрофією (МЛД).
Проблема діагностики та лікування орфанних захворювань є важливим викликом для сучасного суспільства. Так, P.A. Engel і співавт. [1] звертають увагу на те, що середній час від появи перших симптомів орфанного захворювання до встановлення точного діагнозу – близько 4,8 років.
У цій публікації ми наводимо власний досвід діагностики двох різних форм лейкодистрофій – фатальних орфанних захворювань центральної нервової системи (ЦНС): хвороби Канаван та МЛД, кожна з яких має власні особливості клінічного перебігу, результатів нейровізуалізації, біохімічних і генетичних тестів, а також сучасні дані світової літератури про ці захворювання.
Особливістю вказаних захворювань є те, що вони перебігають у дитячому віці під масками таких станів, як гідроцефалія, макроцефалія, дитячий церебральний параліч або психіатричні захворювання (зокрема, шизофренія), однак мають злоякісний прогресуючий перебіг і несприятливий кінець.
Лейкодистрофії є групою генетично детермінованих захворювань, що характеризуються переважним ураженням білої речовини нервової системи. Більшість лейкодистрофій мають тяжкий прогресуючий перебіг і фатальний кінець, причому їх лікування не завжди є ефективним.
З кінця XIX століття у науковій літературі почали з’являтися описи пацієнтів, які мали прогресуючі ураження нервової системи, у яких на аутопсії виявлявся дифузний склероз головного мозку, викликаний дегенерацією білої речовини. Ці захворювання протиставлялися дослідниками розсіяному склерозу, описаному у 1868 р. Ж-М. Шарко. Починаючи з 1899 р. протягом наступних 30 років були описані хвороби Пеліцеуса – Мерцбахера (1899 та 1910 рр.), Краббе (1916 р.), Канаван (1928 та 1931 рр.) та МЛД [2].
Сучасні підходи до класифікації лейкодистрофій засновані на особливостях патогенезу окремих форм, а саме переважного ураження певного компоненту білої речовини [3]. За цим принципом виділяють мієлінопатії, які виникають унаслідок дефекту мієліну або олігодендроцитів (лейкодистрофії з гіпо- або демієлінізацією, лейкодистрофії з вакуолізацією мієліну), астроцитопатії, лейко-аксонопатії, мікрогліопатії та лейко-васкулопатії (табл.)
. 
Новітню класифікацію лейкодистрофій запропонувала данський невролог Marjo van der Knaap, ім’ям якої, зокрема, названа мегаленцефалічна лейкодистрофія з субкортикальними кістами.
Хвороба Канаван
Хвороба Канаван є класичною формою генетично детермінованої лейкодистрофії – мієлінопатії з переважанням у патогенезі вакуолізації мієліну. Хвороба Канаван відома також під такими назвами, як лейкодистрофія Канаван – ван-Богарта – Бертранда, спонгіозна дегенерація ЦНС, інфантильна спонгіозна дегенерація, дефіцит аспартоацилази та N-ацетиласпартатова ацидурія [4].
Уперше це захворювання описали Globus та Straussу 1928 року, а пізніше, у 1931 р., відома американська паталогоанатом Міртель Канаван (1879-1953) (рис. 1) описала летальний випадок захворювання дитини з прогресуючою макроцефалією, у якої на аутопсії була виявлена спонгіозна дегенерація білої речовини головного мозку [5].
 Тільки у 1988 р. було встановлено етіологічний чинник хвороби Канаван, коли Matalonetal і співавт. виявили підвищений рівень N-ацетиласпартату у сечі хворих дітей, а у 1993 р. було виявлено ген-регулятор ферменту аспартоацилази (ASPA), мутації якого викликають це захворювання [6].
Тільки у 1988 р. було встановлено етіологічний чинник хвороби Канаван, коли Matalonetal і співавт. виявили підвищений рівень N-ацетиласпартату у сечі хворих дітей, а у 1993 р. було виявлено ген-регулятор ферменту аспартоацилази (ASPA), мутації якого викликають це захворювання [6].
На сьогодні виділяють дві клінічні форми хвороби Канаван – неонатальну та ювенільну [7].
У більшості випадків спостерігається саме неонатальна, або інфантильна форма хвороби Канаван. Діти народжуються клінічно здоровими, після чого триває період благополуччя, однак у віці 3-5 міс починає розвиватися макроцефалія і затримка психомоторного розвитку.
Затримка розвитку прогресує з віком. У новонароджених порушується функція смоктання, фіксація погляду та контроль за триманням голови. У подальшому у дітей із хворобою Канаван вповільнюється моторний розвиток – відсутні навички сидіння, вставання та ходи, також додається затримка домовленнєвого розвитку у вигляді відсутності гуління та лепету. Відмічаються порушення соціальної взаємодії у вигляді відсутності фіксації погляду та усмішки, однак деякі діти можуть взаємодіяти з оточуючими, усміхатися та слідкувати за предметами. М’язовий тонус на початку захворювання знижений, однак із часом змінюється на спастичний. У хворих дітей розвивається атрофія зорового нерва, що зумовлює відсутність реакції стеження та фіксації погляду. Ураження слуху не характерне.
З віком симптоми захворювання прогресують, у термінальних стадіях пацієнти переважно не можуть рухатися та ковтати, розвиваються епілептичні напади. Прогноз при хворобі Канавана несприятливий, більшість пацієнтів не доживає до другого десятиріччя [8].
На аутопсії зазвичай відмічається субкортикальна спонгіозна дегенерація білої речовини. При електронній мікроскопії виявляються набряклі астроцити, які містять вакуолі та ушкоджені мітохондрії [9].
Залежно від типу генетичної мутації гена ASPА можуть бути різні клінічні прояви хвороби Канаван. Діти з гомозиготною мутацією Tyr231Ter (з відсутністю ферментної активності) клінічно відрізняються від осіб із гомозиготною мутацією Glu285Ala (залишкова ферментна активність) відсутністю різкого прогресування захворювання.
Діти з ювенільною формою хвороби Канаван, як правило, народжуються без ознак захворювання, ростуть та розвиваються з мінімальною затримкою психомоторних навичок. При обстеженні таких дітей часто виявляється підвищений рівень N-ацетил-аспартату в сечі. На МРТ головного мозку за цієї форми захворювання можуть бути виявлені зміни у вигляді підвищеного сигналу в базальних гангліях. Для ювенільної форми хвороби Канаван характерні одиничні мутації (Tyr288Cys, Arg71His або Pro257Arg) із залишковою активністю ферменту аспартоацилази, що зумовлює «м’який» перебіг захворювання. У таких випадках може бути виявлений підвищений рівень ацетиласпартату в сечі та відсутність змін на МРТ головного мозку [10-12].
Мутація виникає в гені ASPA, що бере участь у метаболізмі N-ацетиласпартату. Ця мутація має аутосомно-рецесивний тип успадкування.
Клінічні прояви хвороби Канаван виникають при успадкуванні аномального гена від кожного з батьків за менделевським типом. Якщо людина отримує один нормальний ген і один мутантний, вона буде носієм цієї хвороби, але, як правило, клінічні прояви відсутні.
Ген хвороби Канаван був картований на 17 хромосомі (17pter-P13). Цей ген ASPА кодує аспартоацилазу – фермент, який метаболізує N-ацетиласпарагінову кислоту. N-ацетиласпарагінова кислота є сполукою, яка відіграє важливу роль у функціонуванні білої речовини мозку.
У результаті ураження аспартоацилази відбувається накопичення N-ацетиласпарагінової кислоти в тканині головного мозку. Ознакою хвороби Канаван є аномально високий рівнь N-ацетиласпарагінової кислоти, що може бути виявлений при проведені МР-спектроскопії.
Відповідно до однієї з теорій патогенезу важлива роль відводиться N-ацетиласпарагіновій кислоті як компоненту регуляції водного метаболізму у білій речовині головного мозку. При порушенні функції аспартоацилази виникає хронічний набряк з утворенням кістоподібних порожнин (спонгіозна вакуолізація) у білій речовині мозку, а також утворення аномальних мітохондрій в астроцитах [13, 14].
Хвороба Канаван не має статевої вибірковості, частота ураження осіб чоловічої та жіночої статі є однаковою. Виявлена висока частота носійства мутантного гена у популяції євреїв-ашкеназі (близько 1 на 40-58 осіб). Ризик народження хворої дитини серед євреїв-ашкеназі становить 1 на 6400-13456 випадків. Загальна поширеність і захворюваність на сьогодні не відома [15].
Діагноз в осіб із неонатальною формою хвороби Канаван спирається на показники високої концентрації N-ацетиласпарагінової кислоти (NAA) в сечі. За ювенільної форми хвороби Канаван показник N-ацетиласпарагінової кислоти сечі може бути злегка підвищений.
Отже, аналіз сечі є відносно дешевим та інформативним методом для скринінгової діагностики цього захворювання.
Остаточно для підтвердження діагнозу необхідна молекулярно-генетична діагностика мутації гена ASPA, що кодує фермент аспартоацилазу [16, 17]. Також проводиться визначення активності аспартоацилази у культурі фібробластів, причому зниження її активності підтверджує діагноз навіть за відсутності мутації гена ASPA. Електроенцефалограма може бути корисною у виявленні епілептиформної активності, при цьому фонова активність зазвичай є дифузно уповільненою [8].
Пренатальна генетична діагностика може бути здійснена шляхом вимірювання концентрації N-ацетиласпарагінової кислоти в амніотичній рідині [18]. Проте остаточний діагноз виставляється після проведення молекулярно-генетичного дослідження.
МРТ дозволяє виявити дифузну дегенерацію білої речовини переважно у субкортикальних відділах. Водночас центральні ділянки білої речовини, такі як перивентрикулярна зона та внутрішня капсула, часто залишаються інтактними на початку захворювання. У подальшому до патологічного процесу залучаються бліда куля й таламус, однак хвостате ядро та лушпина не уражуються [19].
На сьогодні ефективного патогенетичного й етіологічного лікування немає, використовується тільки симптоматична терапія. Проте проводяться клінічні випробовування нових методів лікування, описані методики втручання в генетичну поломку гена ASPА для усунення дефектного гена [20]. Інший напрям – це метаболічна терапія з використанням цитрату літію, що має здатність зменшувати рівень N-ацетиласпарагінової кислоти в головному мозку [21]. Також виявлено здатність гліцерил триацетату зменшувати рівень N-ацетиласпарагінової кислоти у мозку хворих мишей і поліпшувати моторні функції. Сьогодні терапія гліцерил триацетатом (у дозі до 4,5 г/кг) використовується при лікуванні дітей, у тому числі новонароджених, із хворобою Канаван [22]. Серед інших метаболічних засобів, ефективність яких зараз досліджується, – тригептатоїн, здатний знижувати оксидантний стрес, збільшувати кількість мієліну в мозку [23].
Переважно застосовується симптоматична терапія у вигляді антиконвульсантів при епілептичних нападах або сечогінних засобів при підвищенні внутрішньочерепного тиску [8].
Клінічний випадок
Хлопчик М., 1 р. 8 міс, поступив у відділення психоневрології нашого інституту зі скаргами матері на наявність у дитини затримки стато-кінетичного і психомовленнєвого розвитку, порушень ковтання та епілептичних нападів в анамнезі.
Дитина народилася від першої вагітності, що перебігала на фоні загрози передчасного переривання на 7 та 16 тижнях гестації, мати отримувала лікування. Зі слів мами, при проведенні ультразвукової діагностики (УЗД) на 23, 36 та 40 тижнях патологічних змін не було виявлено. Пологи термінові, шляхом екстреного кесаревого розтину у зв'язку зі слабкістю пологової діяльності.
Маса при народженні – 3560 г, довжина тіла – 55 см, окружність голови – 37 см, оцінка за шкалою Апгар 7/7 балів. На 3-й день дитина була виписана з пологового будинку.
Перші прояви хвороби виникли у віці 1 міс. У дитини спостерігалися порушення ковтання, підвищена збудливість. Отримував лікування у районній лікарні за місцем проживання з діагнозом синдром м’язового гіпертонусу, афект-респіраторний синдром унаслідок гіпоксично-ішемічного ураження ЦНС. При проведенні нейросонографії (НСГ) виявлено розширення передніх рогів бічних шлуночків, розширення судинних сплетень на рівні каудоталамічної вирізки. Діагностовано страбізм унаслідок парезу окорухового нерва справа. У 5 міс було встановлено затримку хлопчика у розвитку – не тримав голову, не перевертався, не брав іграшки, не фіксував погляд.
Проведена аксіальна комп’ютерна томографія – виявлено ознаки виражених дифузних енцефалопатичних змін, дифузної кортикальної атрофії з переважним ураженням лобної та скроневої частин мозку, помірне симетричне розширення елементів шлуночкової системи мозку. Встановлено діагноз змішаної гідроцефалії, затримки передмовленнєвого та статокінетичного розвитку, псевдобульбарного синдрому. У 8 міс під час гіпертермії з'явилися клоніко-тонічні судоми.
За даними електроенцефалографії (ЕЕГ) виявлено дифузні зміни біоелектричної активності мозку на фоні підвищеної збудливості. В 11 міс встановлено діагноз дитячий церебральний параліч, спастичний тетрапарез, субкомпенсована внутрішня та зовнішня гідроцефалія, псевдобульбарний синдром, симптоматична епілепсія з генералізованими тоніко-клонічними нападами (на фоні терапії депакіном, адренокортикотропним гормоном (АКТГ) напади припинилися).
 Неврологічний статус при надходженні. Дитина притомна, реагує на звернену мову, усміхається. Експресивна мова відсутня. Відмічається вимушена поза – голову не тримає, верхні кінцівки у флексорному гіпертонусі, нижні кінцівки в екстензорному гіпертонусі, не перевертається, самостійне жування відсутнє, ковтає, однак захлинається рідиною. Голова гідроцефальної форми, окружність 52 см (>3SD), велике тім’ячко – 0,5×0,5 см, не напружене, відмічається напруженість потиличних м’язів. Очні щілини симетричні, зіничні рефлекси ослаблені, симетричні. Страбізм справа внаслідок парезу окорухового нерву. За предметом не слідкує. Обличчя симетричне, міміка присутня, відмічається високий рефлекс з м’якого піднебіння, рухи голови обмежені. Тонус м’язів у верхніх і нижніх кінцівках підвищений за пірамідним типом, сухожилкові рефлекси з розширенням рефлексогенних зон, пожвавлені, з елементами клонусу, D=S. Тактильна і больова чутливість збережена. При підтримці опора на передню частину стоп. Крокові рухи не виконує. Функції тазових органів не контролює.
Неврологічний статус при надходженні. Дитина притомна, реагує на звернену мову, усміхається. Експресивна мова відсутня. Відмічається вимушена поза – голову не тримає, верхні кінцівки у флексорному гіпертонусі, нижні кінцівки в екстензорному гіпертонусі, не перевертається, самостійне жування відсутнє, ковтає, однак захлинається рідиною. Голова гідроцефальної форми, окружність 52 см (>3SD), велике тім’ячко – 0,5×0,5 см, не напружене, відмічається напруженість потиличних м’язів. Очні щілини симетричні, зіничні рефлекси ослаблені, симетричні. Страбізм справа внаслідок парезу окорухового нерву. За предметом не слідкує. Обличчя симетричне, міміка присутня, відмічається високий рефлекс з м’якого піднебіння, рухи голови обмежені. Тонус м’язів у верхніх і нижніх кінцівках підвищений за пірамідним типом, сухожилкові рефлекси з розширенням рефлексогенних зон, пожвавлені, з елементами клонусу, D=S. Тактильна і больова чутливість збережена. При підтримці опора на передню частину стоп. Крокові рухи не виконує. Функції тазових органів не контролює.
У соматичному статусі патології не виявлено.
Проведено обстеження: ЕЕГ – виражені дифузні неспецифічні зміни біоелектричної активності головного мозку, уповільнення фонової активності, пароксизмальної епілептичної активності не виявлено. УЗД – ознаки помірних реактивних змін паренхіми печінки. НСГ – підвищення ехогенності мозкової паренхіми, вентрикулодилатація ІІ ст.
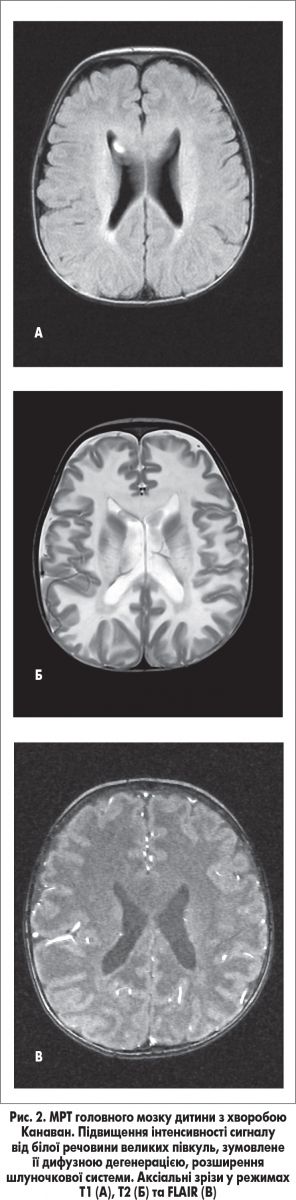
За даними МРТ головного мозку виявлено підвищену інтенсивність сигналу від білої речовини, розширення шлуночкової системи, імовірно, зумовлені захворюванням із групи генетично детермінованих токсичних і метаболічних енцефалопатій (рис. 2). Консультація офтальмолога – виявлено атрофію дисків зорових нервів. Консультативний висновок медичного генетика – у дитини не виключається спадкове дегенеративне захворювання ЦНС: лейкодистрофія Канаван, аутосомно-рецесивний тип успадкування. При молекулярно-генетичному дослідженні ДНК, виділеної з крові пацієнта, у гені ASPА виявлена мутація р. Ala305Glu, що дало можливість остаточно підтвердити діагноз.
Метахроматична лейкодистрофія
Метахроматична лейкодистрофія є типовим захворюванням з ураженням мієліну у вигляді демієлінізації. МЛД – збірна назва гетерогенної групи фатальних захворювань, які характеризуються накопиченням сульфатидів у центральній і периферичній нервовій системі, що призводить до дифузної демієлінізації.
Накопичення метахроматично-реагуючої ліпідної субстанції відбувається у гліальних клітинах, макрофагах, нейронах блідого шару, зубчастого ядра мозочка, ядрах черепних нервів, а також клітинах внутрішніх органів (нирок, жовчного міхура, підшлункової залози, наднирників, печінки). МЛД викликається дефіцитом ферменту арилсульфатази А (ARSA: EC3.1.6.8) та відноситься до групи сфігноліпідозів – лізосомних хвороб накопичення, які характеризуються накопиченням у тканинах органів-мішеней сфігноліпідів. Інші відомі назви захворювання: сульфатидний ліпідоз, синдром Грінфілда, хвороба Шольца, хвороба Геннеберга, синдром Шольца – Більшовскі – Геннеберга [24].
Одну зі своїх назв – хвороба Шольца – МЛД отримала на честь німецького невропатолога та психіатра Вілібальда Оскара Шольца (рис. 3), який у 1925 р. описав сімейний випадок ювенільної форми цього захворювання. У 1928 р. німецькі неврологи Макс Більшовскі (1869-1940) та Ричард Геннеберг (1868-1962) описали те саме захворювання й уперше запропонували термін «лейкодистрофія» для нейродегенеративних захворювань білої речовини. Також у 1928 р. ці дослідники запропонували першу класифікацію лейкодистрофій, засновану на клінічних, патоморфологічних і гістохімічних критеріях.
Зокрема, за гістохімічним критерієм усі лейкодистрофії були поділені на ортохроматичні та метахроматичні. Термін «ортохроматичний» застосовується для опису зразків тканини, які фарбуються в той же колір, що і використовуваний для цього барвник, «метахроматичний» означає фарбування гістологічних структур у колір, невластивий цьому барвнику.
 На честь цих учених ювенільна МЛД отримала одну зі своїх назв – «хвороба Шольца – Більшовскі – Геннеберга» [25]. Макс Більшовскі також є співавтором опису пізньої інфантильної форми нейронального цероїдного ліпофусцинозу, відомого як хвороба Більшовскі – Янського.
На честь цих учених ювенільна МЛД отримала одну зі своїх назв – «хвороба Шольца – Більшовскі – Геннеберга» [25]. Макс Більшовскі також є співавтором опису пізньої інфантильної форми нейронального цероїдного ліпофусцинозу, відомого як хвороба Більшовскі – Янського.
На цей час епонім «хвороба Шольца» майже не використовується, що, на нашу думку, пов’язано з деякими фактами біографії цього дослідника.
Вілібальд Шольц (1889-1971) отримав ступінь доктора 1914 року в Йені, пізніше очолив клініку нервових і психічних хвороб у м. Лейпциг. З 1935 по 1961 рр. він очолював Науково-дослідний інститут психіатрії в м. Мюнхен. Під час Другої світової війни В. Шольц опублікував не менше 11 робіт на основі дослідження мозку 194 осіб – жертв нацистських програм евтаназії «Т‑4», яке проводилося в Науково-дослідному інституті психіатрії м. Мюнхен.
Програма умертвіння «Т‑4» («Акція Тіргартенштрассе 4») – офіційна назва євгенічної програми німецьких нацистів зі стерилізації, а в подальшому і фізичного знищення душевнохворих, розумово відсталих і спадково хворих осіб. В інституті м. Мюнхен його колегою був відомий лікар Юліус Галлеворден, який, використовуючи препарати головного мозку жертв програми «Т‑4», вивчав причини атрофії мозочка, розсіяного склерозу та хорея Гентінгтона.
Відомо, що Галлеворден особисто був присутній при знищенні деяких жертв і самостійно вилучав головний мозок після «евтаназії». Натомість Шольц цікавився переважно пошуками етіології хвороби Літля, психозів і сенільної деменції. У 1956 р. Шольц став головним редактором та автором неврологічного розділу фундаметального «Посібника зі спеціальної патологічної анатомії та гістології», куди увійшли описи деяких його спостережень, зроблені за часів нацизму [25, 27].
У 1964 р. американський невролог Джеймс Остін зі співавторами встановив наявність при МЛД дефекту лізосомального ферменту арилсульфатази А, який призводить до накопичення кислих ліпідів у тканинах пацієнтів і зумовлює ефект метахромазії [28].
Поширеність МЛД становить 1:40 000 – 1:100 000 осіб. Значно вищою є захворюваність у деяких ізольованих популяціях, зокрема у євреїв Хаббана, популяції євреїв, які іммігрували з Йемєну до Ізраїлю, поширеність становить близько 1,3% [29]. Поширеність пізньої інфантильної форми оцінюється у 1:400 000 – 1:170 000 новонароджених [30].
Причиною МЛД є дефіцит ферменту арилсульфатази А, який відповідає за гідроліз сульфатидів, таких як цереброзид‑3-сульфат або 3-О-сульфогалактозілцерамід, до галактоцереброзиду та сульфату. Усього відомі 3 типи арилсульфатази – А, В (лізосомальна) і С (мікросомальна). Патологічне накопичення сульфатидів у нервовій системі (мієліні, нейронах і глії) призводить до ураження білої речовини центральної та периферичної нервової систем, що викликає прогресуючу деменцію, неврологічні відхилення і сліпоту. Метахроматичні гранули, які накопичуються у ЦНС, є високотоксичними, призводять до дегенерації мієліну та втрати аксонів [31].
Дифузна демієлінізація відбувається переважно у півкулях великого мозку, при цьому сіра речовина не уражується. Також відбувається сегментарна демієлінізація периферичних нервів і накопичення метахроматичних гранул у шванівських клітинах, що викликає сенсомоторну поліневропатію, порушення ходи й атаксію.
При МЛД розвивається дифузна симетрична демієлінізація півкуль великого мозку і мозочка в результаті руйнування олігодендроглії, що призводить до аксонального ушкодження, дегенерації пірамідних шляхів з ізоморфним гліозом і демієлінізуючою полінейропатією. Збереженими залишаються лише субкортикальні U-волокна.
Також метахроматичні гранули, які містять сульфатиди, накопичуються в інших тканинах організму, зокрема жовчовивідних протоках печінки, жовчному міхурі, печінці, підшлунковій залозі, яєчниках, лімфатичних вузлах, очах, зубній пульпі та дистальних канальцях нирок.
Підвищений рівень цих сполук також виявляється у сечі [32].
Дефектний ген при МЛД розташований на довгому плечі 22-ї хромосоми (22q13.31-qter) або при недостатності білка-активатора сфігноліпідів – на короткому плечі 10-ї хромосоми (10q21-q22); тип спадкування – аутосомно-рецесивний [33].
Клінічна картина МЛД може бути істотно варіабельною в різних пацієнтів, що пов’язано зі значним поліморфізмом гена ARSA.
Усього до групи метахроматичних лейкодистрофій входять 5 алельних форм.
1. Пізня інфантильна (синдром Грінфілда) – найбільш поширена та тяжка форма, яка відмічається у 50-60% випадків і дебютує в 1-2 роки (частіше у віці 12-18 міс), рідше – у 2-4 роки. Після періоду нормального розвитку починається регрес моторних навичок і мовлення.
Дитина стає дратівливою, погано їсть, порушується хода, з'являється і наростає гіпотонія м'язів, відмічаються часті падіння, нечіткість мовлення, втрата рефлексів (на ранніх стадіях), косоокість, ністагм, мозочкові порушення, атрофія зорових нервів. Перші прояви захворювання можуть з’являтися після перенесеної інфекції або застосування наркозу, після чого зникають на кілька тижнів і потім знову повертаються.
У більшості випадків характерним є швидке прогресування захворювання з приєднанням таких неврологічних ускладнень, як спастичність (у поєднанні з ознаками ураження периферичних нервів), втрата здатності ходити та стояти, епілептичні напади (міоклонічні, парціальні або генералізовані), кіркова сліпота, зниження інтелекту та слуху, псевдобульбарний синдром; на фоні децеребраційної ригідності з опістотонусом настає смерть (зазвичай через 2-4 роки після дебюту).
2. Ювенільна форма (хвороба Шольца) виникає у 20-30% випадків. Дебютує у віці 4-16 років, починаючись із порушень ходи, емоційних і поведінкових розладів. Прояви полінейропатії менш виражені, однак відзначається поява прогресуючої глухоти і сліпоти, афазії, згодом з’являються і посилюються ознаки слабоумства, нетримання сечі та калу, спастичність м'язів, атаксія, мимовільні рухи; іноді виникають судомні напади; середній термін життя складає 7 років, більшість пацієнтів помирають протягом перших 10 років життя, однак відомі випадки, коли тривалість життя таких пацієнтів складала 20 років і більше.
3. Форма дорослих (синдром ван Богарта – Ніссена – Пфайффера) зустрічається рідко. Дебютує у віці 16-60 років. Характеризується повільно прогресуючою деменцією, психозами, атрофією зорових нервів, атаксією, пірамідно-екстрапірамідною симптоматикою, полінейропатією. У літературі є повідомлення про випадки, що проявляються виключно полінейропатією.
4. Часткова недостатність цереброзидсульфатази.
5. Псевдонедостатність арилсульфатази А – синдром, що характеризується вираженим зниженням активності арилсульфатази А за відсутності клінічних проявів.
МЛД може бути запідозреною у пацієнтів із прогресуючими неврологічними порушеннями та з ознаками дегенерації білої речовини за даними МРТ. У типових випадках на МРТ виявляються дифузні симетричні гіперінтенсивні вогнища переважно у тім’яно-потиличних ділянках білої речовини, які згодом поширюються у напрямку лобних ділянок, комісуральних волокон мозолистого тіла та перивентрикулярної білої речовини.
Мірою прогресування захворювання аномалії МРТ стають усе більш вираженими при рострально-каудальній прогресії; розвивається атрофія мозку. Ураження передніх відділів мозку переважно характерне для пацієнтів із пізнім початком захворювання [34].
Гіпоінтенсивні радіальні смужки сигналу між гіперінтенсивними ділянками білої речовини формують МР-симптом «тигрової шкіри» на Т 2-зважених аксіальних зрізах. Збережені периваскулярні ділянки білої речовини перивентрикулярних зон і напівовального центру формують гіпоінтенсивні плями, які нагадують шкіру леопарда [35].
Важливим для встановлення діагнозу є виявлення зниження активності ARSA менше ніж на 10% від норми у лейкоцитах крові або культурі фібробластів шкіри. Однак при використанні лише оцінки активності ARSA можливі помилки у діагностиці, оскільки трапляються пацієнти з псевдодефіцитом цього ферменту.
При таких станах активність ARSA знижена на рівні 5-20% від норми, проте клінічні прояви MЛД відсутні. Також рівень активності ARSA не може бути використаним як предиктор тяжкості перебігу МЛД, тому що не завжди виражений дефіцит ферменту супроводжується тяжкими клінічними проявами.
Остаточно діагноз МЛД повинен бути підтверджений за допомогою молекулярно-генетичного тестування, яке виявляє мутацію гена, що кодує ARSA. Також можлива верифікація діагнозу шляхом виявлення екскреції сульфатидів з сечею методами шарової хроматографії або мас-спектроскопії, також можливе виявлення метахроматичних ліпідних гранул у біоптатах нервової тканини, що є патогномонічним для МЛД [36].
Сьогодні МЛД вважається невиліковним захворюванням із фатальним перебігом. Існує декілька підходів до терапії, які переважно ще на стадії розроблення, зокрема трансплантація кісткового мозку, введення рекомбінантного ферменту ARSA (препарат для замісної терапії «Метазим») або мезенхімальних стовбурових клітин, застосування варфаріну, а також генна терапія [22].
Клінічний випадок
Хлопець К., 16 р., поступив на обстеження до відділення психоневрології для дітей з перинатальною патологією та орфанними захворюваннями зі скаргами на наявність епілептичних нападів генералізованого характеру, що виникають 1-2 рази на півроку, а також прогресуюче зниження когнітивних функцій.
З анамнезу відомо, що хлопець народився, коли матері було 19 років, від першої вагітності, яка перебігала на фоні раннього гестозу (нудоти) протягом перших тижнів. Пологи термінові, фізіологічним шляхом, ускладнені тугим обвиттям пуповини навколо шиї. Навколоплідні води меконіальні. Маса тіла при народженні – 3850 г, зріст – 54 см, обвід голови – 36 см, обвід грудної клітини – 35 см. За шкалою Апгар отримав 7/8 балів. Закричав одразу, прикладений до грудей відразу.
Виписаний із пологового будинку на 4-ту добу. На 1-шу добу життя був неспокійним. Грудне вигодовування до 1 міс. Ранній розвиток без особливостей: голову тримав у 3 міс, сидів у 6 міс, пішов у 1 рік. У 1-й клас пішов у віці 6 років, на той момент вмів читати, рахувати, писати. Сімейний анамнез не обтяжений, має здорову молодшу сестру.
У віці 8-9 років у дитини почалися порушення логічного мислення та дрібної моторики, замкненість, соціальна дезадаптація, порушення поведінки, важко було виконувати домашні завдання, перестав самостійно розбирати шкільні завдання. Стан погіршився після 13 років – знизилася шкільна успішність, з’явилася байдужість до занять, зниження пам’яті та уваги, забував розклад уроків, дні тижня, назви місяців, відмічалося уповільнення мислення. Поступово приєдналася інфантильна поведінка, у їдальні доїдав та допивав після інших учнів, рився у смітті.
У класі був ізольованим, майже не спілкувався з однолітками, ставився до соціальної ізоляції у класі байдуже. Обстежений в обласній психіатричній лікарні, де встановлено діагноз проста шизофренія. У відділенні був пасивним, байдужим, отримував терапію арипіпразолом, на фоні якого був загальмований. Після виписки з відділення оформлено соціальну допомогу з приводу шизофренії. У подальшому кількаразово отримував курси рисперідону без суттєвого позитивного ефекту.
У жовтні 2015 р. вперше у житті виник генералізований епілептичний напад. Другий епілептичний припадок виник у березні 2016 р., після чого призначено вальпроком 750 мг 2 рази на день. Після проведення МРТ головного мозку був направлений до відділення психоневрології ДУ «ІПАГ НАМН» для уточнення діагнозу. З
а період спостереження МРТ головного мозку проводилася тричі: 2014 р. (1 Т) – МР-картина гіпоплазії мозолистого тіла, постгіпоксичних перивентрикулярних вогнищ гліозу, мовновентрикулярної внутрішньої гідроцефалії; 2015 р. (1,5 Т) – ознаки перивентрикулярної лейкомаляції, гіпоплазії мозолистого тіла, помірно вираженої зовнішньої гідроцефалії; 2017 р. (1,5 Т) – МР-картина дисмієлогенного захворювання, гіпоплазії мозолистого тіла, помірна зовнішня регіонарна гідроцефалія. Після проведення третього МРТ головного мозку був направлений до відділення психоневрології ДУ «ІПАГ НАМН» з діагнозом «Симптоматична епілепсія з вторинно генералізованими нападами. Шизофренія, проста форма. Лейкодистрофія?» для уточнення діагнозу.
Під час огляду загальний стан дитини задовільний. Соматичний статус без істотних відхилень.
Контакту доступний, відповідає на запитання, підтримує формальний діалог. Мислення уповільнене, пам’ять знижена. Знижені когнітивні функції. Обвід голови – 57 см. На шкірі спини та плеча справа значних розмірів пігментний невус. Зіниці округлої форми, симетричні. Фотореакція жива, співдружня. Ністагм відсутній. Мімічна інервація симетрична. Ковтання, фонація не порушені. М’язовий тонус задовільний, симетричний. Хода не порушена. Сухожилкові рефлекси нижніх кінцівок живі, D=S, з верхніх – живі, симетричні. Моторна незграбність, хода з елементами атаксії. Патологічні стопні рефлекси відсутні. Черевні рефлекси живі. Координаторні проби виконує. Розлади чутливості відсутні. Порушень функцій тазових органів немає.
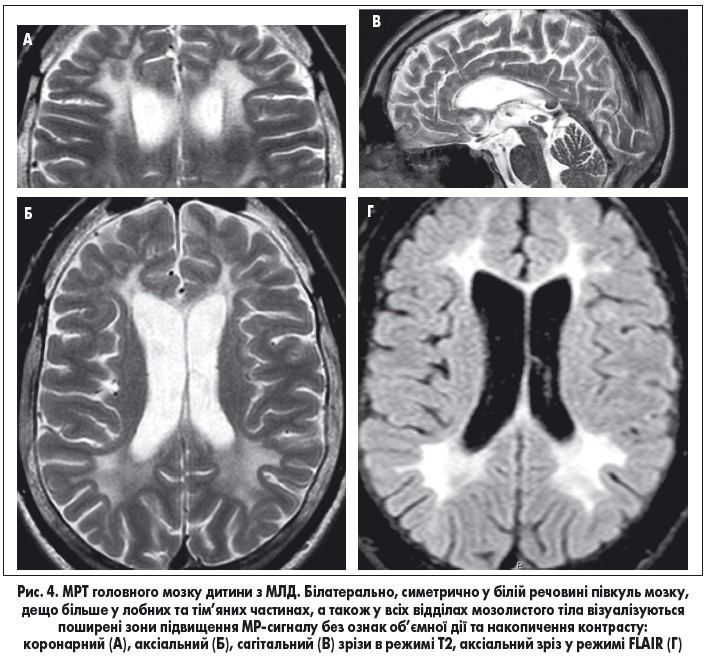
МРТ головного мозку з в/в контрастуванням (рис. 4) – білатерально, симетрично у білій речовині півкуль мозку, дещо більше у лобних і тім’яних частках, а також у всіх відділах мозолистого тіла, візуалізуються поширені зони підвищення МР-сигналу на Т 2 та FLAIR без ознак об’ємної дії та накопичення контрасту. Мозолисте тіло нерівномірно стоншене по всій довжині (мінімально до 2 мм). МР-картина відповідає ознакам метаболічної лейкодистрофії, імовірно, метахроматичної, гіпогенезії мозолистого тіла.
УЗД органів черевної порожнини – реактивні зміни паренхіми печінки.
ЕКГ: синусовий ритм, неповна блокада правої ніжки пучка Гіса. ЕКГ-ознаки ваготонії, частота серцевих скорочень – 62 уд./хв.
ЕЕГ-висновок: епілептиформна активність не виявлена. Виражені зміни біоритміки у вигляді гіперсинхронизації з періодичним уповільненням кіркової ритміки.
Висновок медичного психолога: при огляді орієнтується правильно у місці, часі та власній особистості. Увага нестійка, важко концентрується та переключається, швидко виснажується. Мислення уповільнене, операції аналізу та синтезу ослаблені. Йому складно виділяти основні ознаки та простежувати логічні зв’язки між явищами. Пам'ять знижена переважно за рахунок запам'ятовування. Вербальний інтелект знижений (за даними тесту Векслера). Таким чином, на перший план виступають порушення когнітивних функцій.
При ферментному дослідженні виявлено різке зниження активності арилсульфатази А – 10,2 нмоль/год/мг білка (норма >71 ± 1 нмоль/год/мг білка).
Ураховуючи скарги, анамнез, перебіг захворювання, дані МРТ головного мозку та генетичного дослідження (зниження активності арилсульфатази) встановлено діагноз метахроматична лейкодистрофія, ювенільна форма, аутосомно-рецесивний тип успадкування.
Висновки
Лейкодистрофії, до яких належить згадана вище хвороба Канаван, являють собою одне з найбільш некерованих і прогредієнтних захворювань, що описані в дитячій неврології. Встановлення у дитини цього діагнозу, безумовно, є тяжкою подією не тільки для родини, але і для медичних працівників, що опікуються нею. Рівень розвитку сучасної медицини, на превеликий жаль, не дозволяє відносити це захворювання до курабельних.
Неухильно прогресуючий перебіг з ураженням основних функцій нервової системи з часом призводить до фатальних наслідків. Завданням лікаря є перш за все запідозрити прогресуюче захворювання в дитини з нетиповою картиною органічного ураження нервової системи та своєчасно розпочати діагностичний пошук у цьому напрямі. Встановлення адекватного діагнозу необхідне для родини, щоб позбавити її від марного витрачання часу в пошуках «правильного» діагнозу та проведення необґрунтовано дорогого лікування, а ресурси направити на догляд за дитиною та проведення симптоматичного лікування, яке може істотно подовжити її життя.
Вважаємо за доцільне звернути увагу на те, що нашому пацієнту необхідно було б провести МР-спектроскопію для визначення рівня N-ацетиласпартату в речовині головного мозку, але це дослідження не було проведене у зв’язку з матеріальною неспроможністю батьків (вказане дослідження проводиться в приватних медичних центрах). Хотілося б акцентувати увагу на необхідності створення простору для обміну досвідом між лікарями та родинами пацієнтів, активного їх залучення до обговорення проблем дітей з орфанними захворюваннями на державному рівні.
Література
1. Engel P.A., Bagal S., Broback M., Boice N. Physician and patient perceptions regarding physician training in rare diseases: The need for stronger educational initiatives for physicians. J Rare Dis. 2013; 1: 1-15.
2. Barkovich J. Magnetic resonance neuroimaging. Malden, Mass: Blackwell; 2011.
3. Van der Knaap M.S., Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017 Sep; 134 (3): 351-382.
4. Scriver C.R., Beaudet A.L., Sly W.S., Valle D. The metabolic and molecular bases of Inherited disease, 8nd. New York 2001; 28: 5799-5807.
5. Canavan M.M. Schilder’s Encephalitis PeriaxialisDiffusa. Report of a Case in a Child Aged Sixteen and One-Half Months. Archives of Neurology and Psychiatry. 25 (2): 299-308. doi:10.1001/archneurpsyc.1931.02230020085005.
6. Kaul R., Balamurugan K., Gao G.P., Matalon R. Canavan disease: genomic organization and localization of human ASPA to 17p13-ter and conservation of the ASPA gene during evolution. Genomics. 1994a; 21: 364-70.
7. Breitbach-Faller N., Schrader K., Rating D., Wunsch R. Ultrasound findings in follow-up investigations in a case of aspartoacylase deficiency (canavan disease). Neuropediatrics. 2003; 34: 96-9.
8. Hoshino H. Canavan disease: Clinical features and recent advances in research Pediatrics International (2014) 56, 477-483.
9. Zeng B.J., Wang Z.H., Torres P.A., Pastores G.M., Leone P., Raghavan S.S., Kolodny E.H. Rapid detection of three large novel deletion of the aspartoacylase gene in non-jewish patients with Canavan disease. Mol Genet Metab. 2006; 89: 156-63.
10. Yalcinkaya C., Benbir G., Salomons G.S., Karaarslan E., Rolland M.O., Jakobs C., van der Knaap M.S. Atypical MRI findings in Canavan disease: a patient with a mild course. Neuropediatrics. 2005; 36: 336-9.
11. Kurczynski T.W., Victorio M.C. Atypical Canavan disease associated with a p.A305E mutation and a p.P257R novel variant in the aspartoacylase gene. Abstract 312. Vancouver, Canada: American College of Medical Genetics Annual Clinical Genetics Meeting; 2011.
12. Michals K., Matalon R. Canavan disease. In: Raymond GV, Eichler F., Fatemi A., Naidu S., eds. Leukodystrophies. London: Mac Keith Press, 2011: 156-69.
13. Matalon R.M., Michals-Matalon K. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Front Biosci. 2000; 5: D307-11.
14. Kaul R., Balamurugan K., Gao G.P., Matalon R. Canavan disease: genomic organization and localization of human ASPA to 17p13-ter and conservation of the ASPA gene during evolution. Genomics. 1994a; 21: 364-70.
15. Sugarman E.A., Allitto B.A. Carrier testing for seven diseases common in the Ashkenazi Jewish population: implications for counseling and testing. Obstet Gynecol. 2001; 97: S38-S39.
16. Traka M., Wollmann R.L., Cerda S.R., Dugas J., Barres B.A. PopKo B2008Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci 2011; 537-49.
17. Michals K., Matalon R. Canavan disease. In: Raymond G.V., Eichler F., Fatemi A., Naidu S., eds. Leukodystrophies. London: Mac Keith Press, 2011: 156-6.
18. Al-Dirbashi O.Y., Kurdi W., Imtiaz F., Ahmad A.M., Al-Sayed M., Tulbah M., Al-Nemer M., Rashed M.S. Reliable prenatal diagnosis of Canavan disease by measuring N-acetylaspartate in amniotic fluid using liquid chromatography tandem mass spectrometry. PrenatDiagn. 2009; 29: 477-80.
19. Van der Knaap M.S. Canavan Disease, Magnetic Resonance of Myelination and Myelin Disorders, 3rd edn. Springer, Berlin, 2005; 326-33.
20. McPhee S.W., Janson C.G., Li C., Samulski R.J., Camp A.S., Francis J., Shera D., Lioutermann L., Feely M., Freese A., Leone P. Immune responses to AAV in a phase I study for Canavan disease. J Gene Med. 2006; 8: 577-88.
21. Assadi M., Janson C., Wang D.J., Goldfarb O., Suri N., Bilaniuk L., Leone P. Lithium citrate reduces excessive intra-cerebral N-acetyl aspartate in Canavan disease. Eur J Paediatr Neurol. 2010; 14: 354-9.
22. Anikster Y., Zevin S. et al. A safety trial of high dose glyceryl triacetate for Canavan disease. Mol. Genet. Metab.2011; 103: 203-6.
23. Francis J.S., Markov V., Leone P. Dietary triheptanoin rescues oligodendrocyte loss, dysmyelination and motor function in the nur7 mouse model of Canavan disease. J. Inherit. Metab. Dis. 2014; 37: 369-81.
24. Metachromatic Leukodystrophy Clinical, Biological and Therapeutic Aspects. Latest Findings in Intellectual and Developmental Disabilities Research [Internet]. InTech; 2012 [cited 12 February 2012]. p. 251-360. Available from: http://cdn.intechopen.com/pdfs/28171/InTech-Metachromatic_leukodystrophy_clinical_biological_and_therapeutic_aspects.pdf
25. Zeidman L. RE: Neuroscience in Nazi Europe Part I: Eugenics, Human Experimentation, and Mass Murder. The Canadian Journal of Neurological Sciences. 2012; 39 (03): 400.
26. Bazelon E. Nazi Science Is Still Haunting Anatomy and Fueling Conservatives’ Worst Anti-Abortion Arguments [Internet]. Slate Magazine. 2017 [cited 19 November 2017]. Available from: http://www.slate.com/articles/life/history/2013/11/nazi_anatomy_history_the_origins_of_conservatives_anti_abortion_claims_that.html?src=longreads
27. Kolodny E.H., Fluharty L. Metachromatic leukodystrophy and multiple sulfatase deficiency: sulfatide lipidosis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, 1995, pp. 2693-739.
28. AUSTIN J. Metachromatic Form of Diffuse Cerebral Sclerosis. Archives of Neurology. 1966; 14 (3): 259.
29. MLD 101: Genetics. www.mldfoundation.org. January 6, 2017. Retrieved January 6, 2017.
30. Turpin J.C., Gray F., Baumann N. Leucodystrophies. Encyclopedie MedicoChirurgicale, Neurologie, Elsevier, 1994, 17-076-D-10. Paris.
31. Deconinck N., Messaaoui A., Ziereisen F. Metachromatic leukodystrophy without arylsulfatase A deficiency: a new case of saposin-B deficiency. Eur J Paediatr Neurol, 2008. Vol.12, pp. 46-50.
32. Adam M., Ardinger H., Pagon R. GeneReviews. University of Washington, Seattle; 2017.
33. Горовенко Н.Г., Пічкур Н.О., Ольхович Н.В. Метахроматична лейкодистрофія: особливості діагностики, лікування та медико-генетичного консультування: Методичні рекомендації. – Київ, 2010. – 23 с.
34. Colsch B., Afonso C., Turpin J.C. Sulfogalactosylcer amides in motor and psycho-cognitive adult metachromatic leukodys trophy: relations between clinical, biochemical analysis and molecular aspects. Biochim Biophys Acta, 2008. Vol.1780, pp. 434-40.
35. Krivit W., Saphiro E., Peters C. Adult metachromatic leukodystrophy treated bybone marrow transplantation in 18 patients. SSIEM 39th Annual SymposiumPargue, Czech Republic, 4-7 september. J Inheri Dis, 2001. Vol. 24, p. 103.
36. Martino S., Consiglio A., Cavalieri C., Tiribuzi R., Costanzi E., Severini G.M., Emiliani C., Bordignon C., Orlacchio A. Expression and purification of a human, soluble Arylsulfatase A for Metachromatic Leukodystrophy enzyme replacement therapy. J Biotechnol. 2005 May 25; 117 (3): 243-51.
37. Dali C., Lund A.M. Therapie enzymati que substitutive par intraveineuse pour la leukodystrophie metachromatique (MLD). Congres«Annual Clinical Genetics Meeting», Tampa, Floride (USA), 2009. 25-29 mars.
Тематичний номер «Педіатрія» №1 (44), березень 2018 р.