


9 липня, 2018
Бар’єрна дисфункція при шкірних формах алергії
Шкіра покриває всю поверхню тіла людини, захищаючи її від різноманітних зовнішніх впливів. Порушення епідермального бар’єра призводить до посилення проникнення крізь нього антигенів довкілля та створює умови для розвитку запалення. Ці ланки патогенезу зумовлюють взаємодію зовнішніх антигенів з імунними клітинами організму, що може спричинити системну імунну відповідь [1].
Подібний шлях називається гіпотезою «ззовні всередину» (outside-to-inside), яка пояснює зв’язок між дисфункцією шкірного бар’єра (ШБ) та підвищеним ризиком розвитку алергічних хвороб включно з атопічним дерматитом (АД), бронхіальною астмою (БА), харчовою алергією та алергічним ринітом [2, 3]. Тривале запалення шкіри, у свою чергу, спричиняє подальше ослаблення ШБ, що дозволяє припустити наявність хибного кола за участю ШБ та імунної системи (так звана гіпотеза ззовні всередину та знову назовні, outside-to-inside-and-back-to-outside) [4, 5]. Ці спостереження свідчать, що підтримання функції ШБ є важливим не тільки для ефективного лікування алергічних хвороб, а й для запобігання їх розвитку.
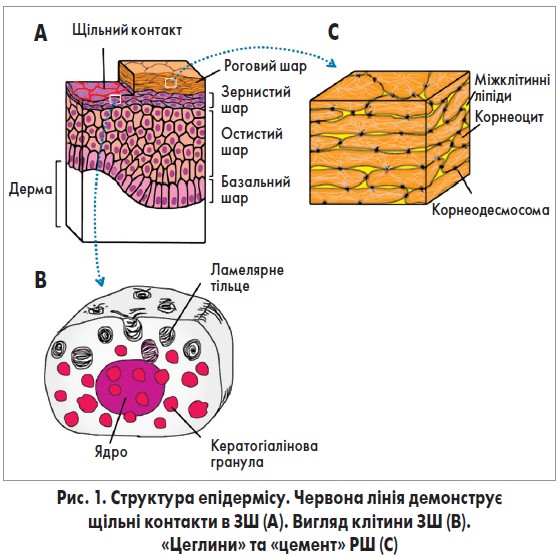 Бар’єрна функція шкіри передусім залежить від стану та функціонування рогового шару (РШ) – зовнішнього шару епідермісу (рис. 1А). РШ утворюється в результаті кератинізації – суворо регульованих процесів диференціації кератиноцитів [6].
Бар’єрна функція шкіри передусім залежить від стану та функціонування рогового шару (РШ) – зовнішнього шару епідермісу (рис. 1А). РШ утворюється в результаті кератинізації – суворо регульованих процесів диференціації кератиноцитів [6].
Під час кератинізації кератиноцити проходять крізь чотири шари епідермісу: базальний, шипуватий, зернистий шар (ЗШ) та власне роговий. У ЗШ кератиноцити починають продукувати два різновиди оточених мембраною гранул: кератогіалінові гранули та ламелярні (пластинчасті) тільця (рис. 1В).
Кератогіалінові гранули містять внутрішньоклітинні компоненти РШ (філагрин, лорікрин, кератинові волокна), у той час як ламелярні тільця включають позаклітинні складники (ліпіди, корнеодесмозин, калікреїни).
У РШ кератиноцити втрачають ядро, стають пласкими і перетворюються на корнеоцити.
Одночасно клітинні мембрани корнеоцитів заміщуються специфічними бар’єрними структурами – так званим роговим конвертом (РК). У процесі руху від ЗШ до РШ ламелярні тільця виходять у міжклітинний простір між корнеоцитами та заповнюють його ліпідами. Ці структури часто характеризують як «цеглини» (корнеоцити) та «цемент» (міжклітинні ліпіди) (рис. 1С).
Утворення бар’єра РШ включає п’ять основних етапів, кожен з яких певним чином пов’язаний з розвитком дерматологічних форм алергії: метаболізм філагрину; формування РК; утворення міжклітинних ліпідів; функціонування корнеодесмосом; злущування корнеоцитів.
Метаболізм філагрину
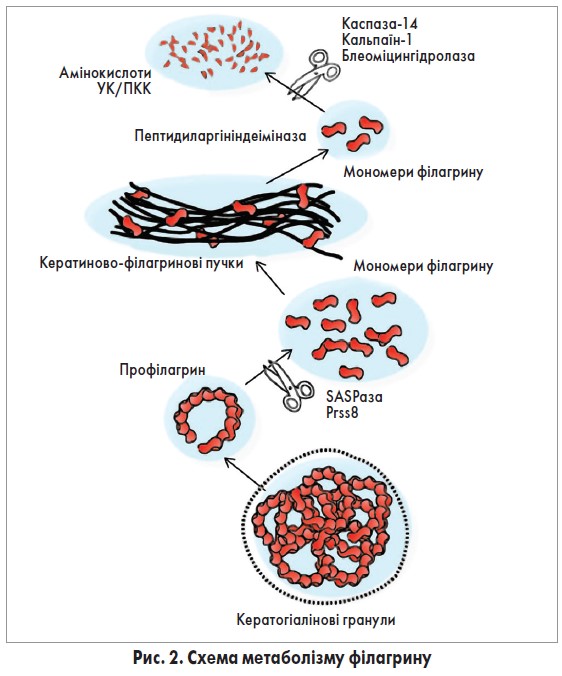 Філагрин та його метаболіти є ключовими речовинами для підтримання нормальної бар’єрної функції шкіри (рис. 2) [7, 8]. У ЗШ філагрин продукується у вигляді полімеру профілагрину, у молекулі якого сполучені 10-12 витків відповідного мономеру. Профілагрин зберігається в кератогіалінових гранулах, а в процесі переходу від ЗШ до РШ розщеплюється з утворенням мономерів за допомогою протеаз CAP1/Prss8 та SASPази/ASPRV1 [9, 10]. Далі мономери філагрину зв’язуються з волокнами кератину; ці пучки є фундаментальною структурою корнеоцитів. У верхніх рівнях РШ філагрин знову вивільняється зі зв’язків з кератином та підлягає подальшому метаболізму, у процесі якого найважливішим є цитрулінування філагрину та кератину за допомогою ферменту пептидиларгініндеімінази [11].
Філагрин та його метаболіти є ключовими речовинами для підтримання нормальної бар’єрної функції шкіри (рис. 2) [7, 8]. У ЗШ філагрин продукується у вигляді полімеру профілагрину, у молекулі якого сполучені 10-12 витків відповідного мономеру. Профілагрин зберігається в кератогіалінових гранулах, а в процесі переходу від ЗШ до РШ розщеплюється з утворенням мономерів за допомогою протеаз CAP1/Prss8 та SASPази/ASPRV1 [9, 10]. Далі мономери філагрину зв’язуються з волокнами кератину; ці пучки є фундаментальною структурою корнеоцитів. У верхніх рівнях РШ філагрин знову вивільняється зі зв’язків з кератином та підлягає подальшому метаболізму, у процесі якого найважливішим є цитрулінування філагрину та кератину за допомогою ферменту пептидиларгініндеімінази [11].
Вивільнені мономери філагрину деградують до вільних амінокислот, у т. ч. глутаміну, аргініну та гістидину, а далі перетворюються на уроканову (УК) та піролідинкарбоксильну (ПКК) кислоти. Цей процес опосередкований іншими протеазами, серед яких каспаза‑14, кальпаїн‑1 та блеоміцингідролаза [12, 13]. УК є важливим хромофором РШ, що абсорбує ультрафіолет та бере участь у підтриманні кислого pH шкіри [14].
Нещодавнє дослідження показало, що УК достовірно знижує експресію костимуляторних молекул на дендритних клітинах та збільшує їх спроможність індукувати формування регуляторних Т-лімфоцитів [15]. У свою чергу, ПКК є одним з головних складників природного зволожуючого фактора, що відповідає за утримання води у РШ. Таким чином, філагрин та його метаболіти відіграють значну роль у бар’єрній функції РШ. Генетичні дослідження встановили, що в мишей з дефіцитом гена філагрину відзначається погіршення функції ШБ та більш виражена сенсибілізація до різноманітних алергенів [16]. Більше того, в штучно виведеної проалергічної лінії мишей BALB/c за умов дефіциту гена філагрину розвивається спонтанний дерматит [17].
Атопічний дерматит та філагрин
АД є найбільш поширеним запальним захворюванням шкіри, що характеризується поліетіологічною природою. Протягом останнього десятиліття результати експериментальних та клінічних досліджень, присвячених цій хворобі, підкреслюють первинну патогенетичну роль порушення функції ШБ [18, 19, 20]. Зокрема, мутації гена філагрину, що супроводжуються втратою функції цієї речовини, зумовлюють розвиток АД та іхтіозу [18, 21].
У північно-європейській та азійській популяціях поширеність мутацій гена філагрину у хворих з АД варіює від 25 до 50% [22, 23]. До того ж геномні асоціативні дослідження (GWAS), проведені серед представників різних рас, ідентифікували 31 локус, пов’язаний з підвищеним ризиком АД, та встановили, що мутація гена філагрину є найпотужнішим серед них чинником ризику [24]. Ці спостереження свідчать про важливу роль дефіциту філагрину в патогенезі АД.
Хоча мутації гена філагрину досить поширені серед мешканців Північної Європи та азійського регіону, вони рідше спостерігаються в Південній Європі та навіть відсутні в деяких країнах Африки [25, 26, 27]. Однак нещодавнє дослідження виявило, що експресія іншого протеїну ШБ – філагрину‑2 – знижена в шкірі пацієнтів з АД, а нонсенс-мутація в гені філагрину‑2 асоціюється зі стійким АД серед представників негроїдної популяції [28, 29]. Біологічна функція філагрину‑2 потребує подальшого з’ясування, однак структура, схема експресії та біологічні властивості цього білка надзвичайно подібні до філагрину. Таким чином, філагрин‑2 також може відігравати важливу роль у підтриманні цілісності ШБ. Слід зазначити можливість того, що дефіцит філагрину може бути компенсованим в умовах тропічного клімату [30].
Утворення рогового конверту
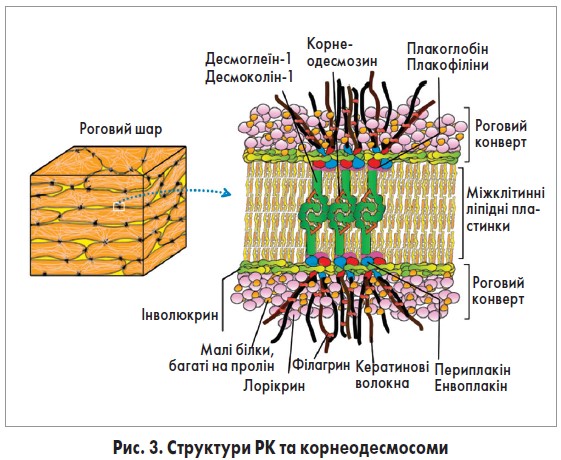
РК є специфічною бар’єрною структурою, що заміщує клітинні мембрани корнеоцитів (рис. 3) [31]. РК складається з тісно зв’язаних нерозчинних білків та позаклітинних ліпідів, прикріплених до них. Ця структура виконує важливу роль фізичного бар’єра РШ.
Формування РК розпочинається у верхніх рівнях остистого шару. У відповідь на підвищення внутрішньоклітинного рівня Ca2+ кератиноцити продукують енвоплакін, периплакін та інволюкрин. Енвоплакін та периплакін утворюють гетеродимери і разом з інволюкрином накопичуються під плазматичною мембраною [32]. Ці три білки зв’язуються за допомогою трансглутамінази (ТГ)-1 та -5 [33].
Інволюкрин виконує роль каркаса РК, у той час як плакінові димери є точками прикріплення кератинових волокон та сполучення їх з білками десмосом. Важливо, що, оскільки плакінові білки щільно прилягають до інволюкринового каркаса, десмосоми та кератинові волокна утворюють жорсткі зв’язки з РК, що забезпечує механічну стабільність корнеоцитів.
У ЗШ продукуються лорікрин та малі білки, багаті на пролін. Ці білки «зшиваються» за допомогою ТГ‑3 та потрапляють на периферію клітин, де прикріплюються до інволюкринового скелета під дією ТГ‑1 та ТГ‑5 [34]. З метою зміцнення РК це «зшивання» повторюється кілька разів, внаслідок чого вміст лорікрину зростає приблизно до 80% усіх білків РК. ТГ‑1 також бере участь у вбудовуванні позаклітинних ліпідів керамідної природи в інволюкриновий скелет та заміщенні керамідами подвійного ліпідного шару плазматичної мембрани [35].
Утворення РК та дерматологічні форми алергії
Незважаючи на повсюдну присутність інволюкрину, енвоплакіну та периплакіну в РК, у мишей з нокаутом (відсутністю) гена будь-якого із цих білків не відзначається очевидних аномалій шкіри [36, 37, 38]. Нокаут усіх цих генів одночасно призводить до порушень утворення РК, зокрема, зниженого вмісту ліпідів та погіршеної механічної цілісності, однак функція ШБ зберігається (імовірно, компенсується шляхом зменшення лущення корнеоцитів) [39].
Аналогічно миші з дефіцитом лорікрину відрізняються тільки фенотипом: блискучою шкірою при народженні та зниженою стабільністю РК [40]. Ці дослідження дозволяють припустити, що білки РК містяться в організмі в надлишку, а формування РК має потужні компенсаторні механізми. Це підтверджується тим, що жодні мутації генів, які кодують утворення та функції компонентів РК, не асоціюються з патогенезом дерматологічних форм алергії.
РК є порушеним або навіть відсутнім за умов дефіциту ТГ‑1, при якому розвивається тяжка іхтіозіформна еритродермія (автономний рецесивний вроджений іхтіоз 1 – АРВІ 1) [41]. До того ж дефіцит ТГ‑5 спричиняє розвиток пілінг-синдрому, що проявляється на межі зернистого та рогового шарів [42]. Ці факти свідчать про суттєву роль ТГ в утворенні РК; однак про зв’язок між генетичними мутаціями ТГ та алергічними хворобами шкіри раніше не повідомлялося.
Утворення міжклітинних ліпідних пластинок
 Міжклітинні ліпіди (т. зв. цемент) є також важливим компонентом бар’єра РШ (рис. 3). Вони є гетерогенною сумішшю керамідів, вільних жирних кислот та холестерину в приблизному молярному співвідношенні 1:1:1. Ці ліпіди продукуються в ЗШ, зберігаються в ламелярних тільцях, а в подальшому виділяються в позаклітинний простір у процесі руху клітин до РШ.
Міжклітинні ліпіди (т. зв. цемент) є також важливим компонентом бар’єра РШ (рис. 3). Вони є гетерогенною сумішшю керамідів, вільних жирних кислот та холестерину в приблизному молярному співвідношенні 1:1:1. Ці ліпіди продукуються в ЗШ, зберігаються в ламелярних тільцях, а в подальшому виділяються в позаклітинний простір у процесі руху клітин до РШ.
Тільки керамідна фракція ліпідів РК містить понад 300 окремих видів сполук [43]. Серед них незамінним є омега-гідроксикерамід, оскільки він з’єднується з інволюкриновим каркасом під дією ТГ‑1 та покриває поверхню корнеоцитів. Використовуючи цей керамід як своєрідний шаблон, у міжклітинному просторі між корнеоцитами формуються шари ліпідних пластинок [44].
Утворення міжклітинних ліпідів та дерматологічні форми алергії
Деякі дефекти ферментів, що беруть участь у метаболізмі керамідів, пов’язані з етіологією хвороб шкіри, які супроводжуються порушенням її бар’єрної функції. Основними ферментами утворення омега-гідроксикераміду є 12R-ліпоксигеназа, що кодується геном ALOX12B, та епідермальна ліпоксигеназа‑3, що кодується геном ALOXE3 [45]. Дефект цих ферментів спричиняє вроджений іхтіоз (АРВІ2 та АРВІ3 відповідно) [46]. Шкірні прояви АРВІ 2 та АРВІ 3 є менш тяжкими, ніж прояви АРВІ 1, ймовірно, тому, що в разі цих хвороб білковий шар РК все ж таки утворюється.
Трансмембранний транспорт ламелярних тілець відбувається за допомогою білка-переносника – АТФ-зв’язувального касетного транспортера‑12 підродини А (ABCA12) [47]. Мутації гена цього білка призводять до помірного (АРВІ 4А) чи тяжкого вродженого іхтіозу (АРВІ 4В, також відомого як іхтіоз арлекіна), що дозволяє припустити важливу роль вмісту ламелярних тілець у процесі зроговіння.
Нещодавно також було встановлено участь у секреції вмісту ламелярних тілець трансмембранного білка‑79 (маттрину) [48, 49]. У мишей з дефіцитом цього білка розвивається спонтанний дерматит з підвищеним рівнем сироваткового IgE, подібний до АД у людей. До того ж метааналіз досліджень за участю хворих з АД виявив, що міссенс-мутація гена маттрину незначно, але достовірно асоціювалася з АД [49]. Як свідчать дані, порушення функції ламелярних тілець та розлади формування міжклітинного ліпідного шару внаслідок цих порушень можуть призвести до погіршення бар’єрної функції шкіри у хворих з АД.
Структура корнеодесмосом
Клітинна адгезія корнеоцитів залежить від десмосомного апарату – корнеодесмосоми (рис. 3). Десмосома складається з трьох родин білків: десмосомні кадгерини, т. зв. білки-броненосці (armadillo) та плакіни. У корнеодесмосомах десмоглеїн‑1 та десмоколлін‑1 (родина кадгеринів) взаємодіють із плакоглобіном та плакофілінами (білки-броненосці), що, у свою чергу, прикріплюються до енвоплакіну та периплакіну. Як було зазначено вище, гетеродимери енвоплакіну та периплакіну прикріплюються до інволюкринового каркасу та зв’язують кератинові волокна. Іншим важливим модулятором корнеодесмосомальної адгезії є корнеодесмозин, який зберігається в ламелярних тільцях, а далі секретується в міжклітинний простір РШ, де взаємодіє з кадгеринами та бере участь в адгезії [50].
Утворення корнеодесмосом та дерматологічні форми алергії
Порушення корнеодесмосомального апарату супроводжується схильністю до надмірного відлущування корнеоцитів, що може спричинити дефект ШБ та подальше запалення шкіри. Нещодавнє дослідження виявило, що гомозиготна мутація десмоглеїну‑1 призводить до тяжкого дерматиту (еритродермії), який супроводжується долонно-підошовною кератодермією, гіпотріхозом та підвищеним рівнем сироваткового IgE (синдром, відомий також як тяжкий дерматит, множинна алергія та синдром SAM) [51].
Важливо, що в пацієнтів із цим синдромом часто спостерігаються харчові алергії. У свою чергу, гомозиготна мутація корнеодесмозину спричиняє синдром лущення шкіри‑1, що проявляється дерматитом, тяжким свербінням, харчовими алергіями, повторними епізодами ангіонабряку та кропив’янки, БА та підвищеним сироватковим рівнем IgE [52].
Десквамація корнеоцитів
Корнеоцити поверхні РШ постійно відлущуються. Цей феномен називається десквамацією і є вкрай важливим для підтримання гомеостазу РШ. Десквамація корнеоцитів здебільшого регулюється протеолітичним каскадом калікреїнів (КЛК‑5, КЛК‑7, КЛК‑14) [53]. Активність цих ферментів є pH-залежною та посилюється за умов підвищення pH у РШ. Також їх дія суворо регулюється інгібіторами протеаз, зокрема лімфоепітеліальним інгібітором серинових протеаз LEKTI, що кодується геном SPINK5 [54]. КЛК та цей інгібітор зберігаються у ламелярних тільцях та секретуються в міжклітинний простір на межі зернистого та рогового шарів.
Десквамація корнеоцитів та дерматологічні форми алергії
У пацієнтів з АД pH поверхні шкіри збільшений, у т. ч. у зв’язку зі зменшеною продукцією УК з філагрину (рис. 2) [55]. Внаслідок цього за умов АД підвищується активність КЛК, що негативно впливає на бар’єрну функцію РШ кількома шляхами. По-перше, КЛК розщеплюють кадгерини корнеодесмосом та стимулюють відлущування корнеоцитів. По-друге, ці ферменти активують рецептор PAR‑2, розміщений на кератиноцитах. Активація рецепторів PAR‑2 спричиняє гальмування секреції ламелярних тілець шляхом пригнічення ферментів, які беруть участь у переробці ліпідів [56]. І, зрештою, активовані КЛК підвищують продукцію інтерлейкіну (ІЛ)-1α та ІЛ‑1β, попередники яких у великих кількостях містяться в цитозолі корнеоцитів. Підтверджено, що в РШ хворих з АД збільшений рівень ІЛ‑1, підвищена продукція якого асоціюється з дефіцитом філагрину [57].
З патогенезом АД пов’язано два генетичні поліморфізми, які призводять до підвищеної активності КЛК: мутація KЛK‑7, що супроводжується набуттям функції, та мутація SPINK5, що супроводжується втратою функції. SPINK5 відомий як ген, що відповідає за розвиток синдрому Незертона, при якому в пацієнта виникає широкий спектр алергічних проявів, у т. ч. дерматит за типом АД, харчові алергії, астма, сінна лихоманка, значно підвищений рівень сироваткового IgE [60]. Істотну асоціацію поліморфізму SPINK5 з АД було підтверджено в британському та азійському дослідженнях, однак не виявлено у французькому [61, 62, 63, 59].
Щільні контакти при дерматологічних формах алергії
Для цілісності ШБ украй важливою є також наявність щільних контактів (ЩК), які «зшивають» прилеглі кератиноцити РШ (рис. 1А), створюючи бар’єр для проникнення води та розчинених у ній речовин [64]. ЩК складаються з трансмембранних білків, зокрема родин клаудину та окклюдину, а також кількох цитозольних каркасних білків. Незамінну роль ЩК у гомеостазі шкіри було вперше продемонстровано в дослідженні на мишах з дефіцитом клаудину‑1, що загинули впродовж 24 год після народження від тяжкого зневоднення [65].
Важливо, що в цих мишей не спостерігалося аномалій у продукуванні компонентів РК. Нещодавнє дослідження на мишах зі змодельованим АД показало, що запалення шкіри пригнічувало експресію білків ЩК, а дефіцит філагрину на неї не впливав [66].
У людей експресія клаудину‑1 знижена в неуражених ділянках шкіри пацієнтів з АД. Раніше повідомлялося про зв’язок поліморфізму гена клаудину‑1 зі схильністю до АД [67]. Ці спостереження свідчать, що порушення ЩК призводять до бар’єрної дисфункції шкіри, яка спостерігається в пацієнтів з АД.
Оскільки більша частина шкіри покрита РШ, ЩК діють як друга лінія захисту від зовнішніх патогенів; однак у придатках шкіри (волосяні фолікули, потові залози) ЩК виступають первинними бар’єрними структурами, оскільки там відсутній РШ. Відомо, що волосяні фолікули є важливими обхідними шляхами проникнення у шкіру ліків та хімічних речовин [68].
Це підтверджується тим, що широко розповсюджені висипні інфекції, викликані вірусом простого герпесу чи контагіозним молюском, які проникають до організму через волосяні фолікули, іноді розвиваються як ускладнення АД [69, 70]. Отже, в пацієнтів з АД при розладах утворення/функціонування ЩК придатки шкіри є шляхом проникнення патогенів.
Імунологічна модуляція ШБ
Сьогодні продовжують накопичуватися докази стосовно того, що імунні клітини здатні впливати на цілісність шкіри шляхом продукції цитокінів [71, 72]. Хоча вогнища ураження при АД формуються за умов складної взаємодії імунних клітин, імунопатогенез АД передусім характеризується порушеною відповіддю Th2-клітин [73, 74]. Попередні дослідження показали, що ІЛ‑4 та ІЛ‑13, два головних цитокіни Th2-клітин, пригнічують продукцію філагрину та кератинів, компонентів РК (лорікрину та інволюкрину), молекул клітинної адгезії (десмоглеїни) та ліпідів керамідної природи. ІЛ‑31, ще один цитокін Th2-клітин, також пригнічує експресію гена філагрину [75]. Більше того, нещодавнє дослідження виявило, що ІЛ‑33, який у великій кількості накопичується в кератиноцитах, так само здатен інгібувати експресію гена, що кодує філагрин [76].
Ймовірно, першопочатковим призначенням цих імунологічних дій, спрямованих на порушення цілісності шкіри, є сприяння відлущуванню та заміні пошкоджених корнеоцитів, однак сукупність ефектів цих цитокінів може спричинити порушення бар’єрної функції шкіри, особливо в пацієнтів з АД. Хибне коло за участю вродженого порушення ШБ та розладів імунного бар’єра зумовлює формування хронічного стійкого запалення шкіри при АД.
Бар’єрна дисфункція як провідна ланка патогенезу дерматологічних форм алергії
Доведено, що нашкірні антигени є потужними сенсибілізаторами для алергічних хвороб. Дослідження на мишах показали, що харчова алергія та БА можуть бути індуковані шляхом нашкірної сенсибілізації, особливо при порушенні функції ШБ. У людей послідовний розвиток алергічних хвороб (атопічний марш) часто спостерігається при АД та деяких генетичних дерматозах, наприклад, синдромі Незертона, пілінг-синдромі, SAM-синдромі. Це свідчить про те, що порушення бар’єрної функції шкіри бере участь у розвитку атопічного маршу.
Ще одним хронічним імунним розладом, асоційованим з гіперчутливістю до їжі, є еозинофільний езофагіт. Нещодавно було встановлено зв’язок цієї хвороби з мутаціями кальпаїну‑14 (CAPN14) – специфічної протеази клітин стравоходу [77]. У свою чергу, експеримент in vitro показав, що надмірна активація CAPN14 призводить до втрати десмоглеїну‑1 [78].
Ці дослідження демонструють, що порушення бар’єрної функції епітелію слизової оболонки також бере участь в індукції алергічних хвороб. Клінічні дослідження встановили, що нашкірне нанесення антигенів зумовлює сенсибілізацію, у той час як пероральне вживання антигенів індукує імунну толерантність [79, 80].
За наявності порушень бар’єрної функції шкіри чужорідні антигени легко проникають в епідерміс та активують імунні рецептори та рецептори розпізнавання паттернів. Це призводить до продукування цитокінів, що стимулюють Th2-клітини (зокрема, ІЛ‑33, ІЛ‑25), та тимусних стромальних лімфопротеїнів (ТСЛП). Дослідження на тваринах продемонстрували важливу роль ТСЛП в індукції харчової алергії з ураженням шкіри, подібним до АД, при нашкірному нанесенні алергену.
Підвищений рівень ТСЛП в епідермісі спричиняє накопичення базофілів у шкірі та стимулює цитокінову відповідь [81]. До того ж вплив ТСЛП на клітини Лангерганса в епітелії може відігравати важливу роль у продукції IgE під час нашкірної сенсибілізації до харчових алергенів [82]. У дослідженні на імуноскомпроментованих мишах, яким було пересаджено людську шкіру, також було показано супресивну дію ТСЛП на експресію гена філагрину [83].
Слід зазначити, що цитокіновий профіль пацієнтів з іхтіозом, які є носіями мутації гена філагрину, характеризується домінуванням ІЛ‑17 та низьким рівнем експресії цитокінів, пов’язаних з Th2-клітинами [84]. Такий профіль більш характерний для пацієнтів із псоріазом, ніж для хворих з АД.
Терапевтичний підхід до відновлення функціонування ШБ
Недостатнє функціонування ШБ та надмірна імунна відповідь є двома сторонами однієї медалі в патогенезі дерматологічних форм алергії, і одне завжди пов’язане з іншим [85]. Таким чином, для ефективного лікування дерматологічних форм алергії потрібно використовувати засоби, спрямовані на зміцнення ШБ, та імуносупресивні препарати. Нещодавно дві групи дослідників вивчали вплив зміцнення ШБ за допомогою зволожуючих засобів упродовж неонатального періоду на запобігання розвитку АД [86, 87]. Вони встановили, що застосування зволожувачів на ранньому етапі життя супроводжувалося меншою (на 32-50%) частотою розвитку АД. Ці результати свідчать, що уникнення сенсибілізації при нашкірному нанесенні антигенів шляхом зміцнення ШБ у неонатальному періоді є багатообіцяючою стратегією у профілактиці АД.
Також було продемонстровано потенціал філагринозамісної терапії. Цей підхід передбачає застосування препаратів, що пригнічують передчасні стоп-кодони (read-through drugs), та ліків, що посилюють продукцію філагрину, або безпосередньо мономеру філагрину чи його метаболітів. Препарати, які пригнічують передчасні стоп-кодони, здатні запобігати нонсенс-мутаціям гена філагрину, що робить їх вдалим вибором для носіїв як гетеро-, так і гомозиготних мутацій цього гена.
Ймовірними засобами з таким ефектом є антимікробні пептиди (гентаміцин, PTC124 – аталурен), які сьогодні проходять клінічні випробування для лікування інших генетичних захворювань [88, 89]. Своєю чергою, медикаменти, що посилюють продукцію філагрину, підходять тільки для пацієнтів з гетерозиготними мутаціями гена, що кодує цей білок. Ймовірними засобами з подібною дією є агоністи рецепторів, що активуються пероксисомним проліфератором [90], апігенін [92], сечовина [94], ще кілька засобів, що перебувають на стадії розробки [93, 83], багата на серин дієта [91].
Вищеназвані медикаменти продемонстрували здатність індукувати диференціацію кератиноцитів та підвищувати рівні філагрину. Однак існує потреба в подальших дослідженнях, оскільки ефективність цих препаратів була оцінена тільки in vitro або на тваринних моделях.
Інтенсивні дослідження та пошук інноваційних методик поліпшення функції ШБ триває [95]. Отже, можна сподіватися на те, що в найближчому майбутньому лікування шкірних проявів алергії значно покращиться.
Список літератури знаходиться в редакції.
Egawa G., Kabashima K. Barrier dysfunction in the skin allergy.
Allergology international, 2018. Volume 67, Issue 1, P. 3-11.
Переклала з англ. Лариса Стрільчук
Медична газета «Здоров’я України 21 сторіччя» № 11-12 (432-433), червень 2018 р.