


7 вересня, 2015
Порушення статевого розвитку у дівчаток

Содержание статьи:
- Затримка статевого розвитку.
- Синдром Шерешевського – Тернера.
- Чиста форма дисгенезії гонад.
- Змішана форма дисгенезії гонад.
[Продовження. Початок в МАЗЖ, 2015, № 2 (88).]
>>__У статті висвітлено сучасні аспекти проблеми порушення статевого дозрівання у дівчаток. Представлено клінічні прояви основних форм патології, можливості діагностики та лікування.
Ключові слова: статевий розвиток дівчаток, передчасне дозрівання, затримка статевого розвитку.
Затримка статевого розвитку
Затримка статевого дозрівання – це відсутність вторинних статевих ознак у підлітків віком 13-14 років, відсутність менархе впродовж 3 років і більше від початку розвитку вторинних статевих ознак, а також першої менструації до 15-річного віку, невідповідність показників зросту та маси тіла дівчинки хронологічному віку.
За даними різних авторів, у підлітків частота затримки статевого розвитку коливається від 14 до 33 % і в структурі гінекологічної захворюваності становить 2-7 %.
Затримка темпів статевого розвитку – поява вторинних статевих ознак та менархе у фізіологічні терміни з подальшим порушенням встановлення менструального циклу протягом 2 років і більше, а також з повільним розвитком вторинних статевих ознак чи призупиненням їх розвитку. У залежності від термінів затримки встановлення менструального циклу виділяють 3 ступені: І ступінь – 2 роки; II – 3 роки; III ступінь – 4 роки і більше.
У повсякденній роботі лікаря зручною є класифікація, що представлена у таблиці 2.
Таблиця 2. Практична класифікація варіантів затримки статевого розвитку
|
Генетично зумовлена група(характерний гіпергонадотропний стан) |
Сімейні варіанти, хромосомні захворювання,генна патологія |
|
Група центрального генезу (характерний стан гіпогонадотропного гіпогонадизму) |
Вроджені чи набуті ураження гіпоталамуса та гіпофіза |
|
Група периферійного генезу (характерний гіпергонадотропний стан) |
Недостатність чи втрата яєчників, рефрактерність статевих органів |
|
Група соматогенного генезу |
Синдром мальабсорбції, хронічна ниркова патологія, ендоінтоксикація, недостатність ендокринних залоз, аліментарний фактор, анемії |
У дівчат із затримкою статевого розвитку можливі такі клінічні прояви:
- кольорова сліпота;
- аносмія;
- носові кровотечі;
- приливи;
- дизурія;
- відсутність інтересу до протилежної статі;
- інфантильний та астенічний морфотипи;
- нормоскелія (реєструється у 5 разів рідше, ніж серед популяції);
- гіпоестрогенія;
- статевий інфантилізм;
- гіпотрофія зовнішніх статевих органів;
- первинна чи вторинна аменорея;
- альгоменорея;
- ювенільні кровотечі.
Затримка статевого розвитку центрального генезу
Гіпоталамічний гіпогонадотропний гіпогонадизм (гіпогонадотропний євнухоїдизм, синдром Кальмана, або ольфактогенітальний синдром). Етіологічним фактором захворювання є порушення функції гіпоталамічних структур під впливом спадкових чинників чи інфекцій та інтоксикацій, перенесених у ранньому дитячому віці. Як наслідок, виникають порушення, пов’язані з ізольованою гіпоталамічною недостатністю, дефектом розвитку центру нюху.
Спадково зумовлені форми гіпогонадотропного гіпогонадизму
Синдром Лоренса – Муна – Барде – Бідля. Рівень захворюваності становить один випадок на 60 000 пацієнток. Захворювання має сімейний характер, що пов’язано з множинними дефектами гена, дегенеративними змінами ядер гіпоталамуса, зменшенням гангліозних клітин з розростанням на їх місці клітин глії.
Клінічна картина проявляється гіпогонадизмом, розумовою відсталістю, пігментним ретинітом, аномаліями розвитку кисті, ожирінням.
Лікування: дотримання дієти з обмеженням жирів, вуглеводів, висококалорійних страв, лікувальна фізкультура, гормональна терапія (тиреоїдин по 0,05-0,2 г/доб протягом 5 днів з перервою на 2-3 дні, гонадотропні гормони, циклічна терапія статевими стероїдними гормонами упродовж 12-13 років).
Прогноз захворювання не досить сприятливий – розумова відсталість не зникає, на фоні деякого поліпшення статевого розвитку зменшується ожиріння, однак менструальний цикл не відновлюється.
Хвороба Хенда – Шюллера – Крісчена. Це захворювання має аутосомно-рецесивний тип успадкування і пов’язане з ураженням гіпоталамо-гіпофізарної системи внаслідок порушення обміну ліпідів. У результаті метаболічних розладів виникають гістіоцитарні гранульоми в різних органах, у т.ч. і в гіпоталамусі.
Для клінічної картини характерні нанізм, статевий інфантилізм, екзофтальм, нецукровий діабет, ксантоматоз, збільшення лімфатичних вузлів, виражені зміни кісткового скелета.
Лікування: дотримання дієти з обмеженням ліпідів; можлива ефективність рентгенотерапії на гіпофізарно-діенцефальну ділянку і вогнища грануляційної тканини.
Гіпофізарний гіпогонадотропний гіпогонадизм
Етіологія і патогенез цієї форми затримки статевого розвитку пов’язані з органічними ураженнями гіпофіза внаслідок його природженої гіпоплазії або гіпоксії. Зазвичай нестача кисню виникає в постнатальному періоді в результаті захворювань, які порушують кровообіг у передній частці гіпофіза, або після стиснення його (синдром «порожнього» турецького сідла).
Синдром «порожнього» турецького сідла – комплекс ендокринних, неврологічних та офтальмологічних порушень, що виникає внаслідок зниженої або «німої» функції гіпофіза. За етіологією синдром поділяється на первинний і вторинний. При первинному спостерігається стиснення та сплощення гіпофіза грижею павутинної оболонки головного мозку, яка проникає в інтраселярну ділянку через отвір ніжки гіпофіза в діафрагмі турецького сідла. При вторинному синдромі гіпофункція гіпофіза може бути спричинена стисненням та зруйнуванням пухлиною його передньої частки.
Розміри та контури турецького сідла не змінені, однак гіпофізарна ямка на рентгенограмі виглядає порожньою. Пангіпопітуїтаризм (функціональна неспроможність усієї передньої частки гіпофіза) призводить до вираженої затримки росту та статевого дозрівання, інфантилізму, первинної аменореї. Біохімічні зміни полягають у вибірковому порушенні синтезу β-ланцюгів (ізольована недостатність ФСГ та ЛГ) або α-ланцюгів гонадотропінів (недостатність продукції ФСГ, ЛГ, ТТГ), що клінічно проявляється вторинним гіпотиреозом. Пацієнтки скаржаться на частий головний біль, відсутність менструацій та затримку статевого розвитку.
Гармонійний фізичний розвиток відзначається тільки у 25 % пацієнток із затримкою статевого розвитку центрального генезу. При пангіпопітуїтаризмі спостерігається низький зріст, однак за відсутності порушень вмісту соматотропного гормона (СТГ) можуть зберігатися вікові зростові параметри. Затримка статевого розвитку зумовлена низькою секрецією гонадотропінів і, як наслідок, порушенням розвитку фолікулярного апарату яєчників, зниженням вироблення естрогенів. Проявом гормональної дисфункції є атрофічний тип піхвових мазків.
Слід зазначити, що соматичні аномалії відсутні, будова тіла пропорціональна, не спостерігаються або слабко розвинуті вторинні статеві ознаки, наявна гіпоплазія зовнішніх і внутрішніх статевих органів. При цитогенетичному обстеженні хворих завжди виявляється жіночий каріотип (46,XX).
У діагностиці вирішальне значення має рентгенографія черепа, при якій виявляють ознаки підвищеного внутрішньочерепного тиску (посилення пальцевих втиснень у склепінні кісток черепа), пошкодження в ділянці турецького сідла (широкий вхід, зміна розмірів, форми тощо). Зміни на енцефалограмі свідчать про гіпоталамічну недостатність, дисфункцію підкіркових структур, значно рідше – органічне ураження головного мозку. Ознаки патології виявляють і під час огляду неврологом.
Секреція ЛГ та ФСГ знижена до 1,0-3,0 мМО/мл та нижче. Відзначається низька секреція естрогенів. Характерні високий вміст серотоніну в крові (> 0,6 мкмоль/л), зниження екскреції норадреналіну < 70,0 нмоль/доб та підвищення секреції мелатоніну > 50,0 нмоль/доб. Ехографічно візуалізуються гіпопластична матка та маленькі яєчники зі слабко вираженим фолікулярним апаратом.
Ступінь функціональних можливостей яєчників оцінюють за результатами проби з пергоналом під контролем УЗД, секреції естрогенів, лапароскопії. Для проведення проби пергонал-500 дозою 150 МО/доб вводять внутрішньом’язово протягом трьох днів, а надалі дозою 225-300 МО/доб (максимальна кількість препарату 25 ампул). На позитивну пробу вказують поява симптому «зіниці», підвищення еластичності цервікального слизу, рівня естрадіолу в крові до 500-600 нг/л, збільшення об’єму яєчників та фолікулярного апарату. Якщо вищеперераховані ознаки після введення пергоналу не виявляються, це свідчить про відсутність фолікулярного апарату в яєчниках. Остаточний діагноз встановлюють після біопсії гонад.
Для виключення ураження передньої частки гіпофіза проводять пробу з гонадоліберином. Одномоментно внутрішньовенно вводять 100 мкг гонадоліберину. Моніторинг секреції ЛГ та ФСГ здійснюють до введення препарату, а також кожні 15 хв протягом 2 год після введення. Підвищення секреції ЛГ та ФСГ у більш ніж 5 разів свідчить про збережену гонадотропну функцію гіпофіза і недостатню секрецію гонадоліберину.
Лікування синдрома «порожнього» турецького сідла має бути комплексним, його проводять спільно з терапевтом, невропатологом, ендокринологом.
При затримці статевого розвитку, пов’язаній з дефіцитом маси тіла, гормональна терапія протипоказана. Рекомендовано нейротропні препарати, симптоматичну терапію, вітамінотерапію, психотерапію, збалансоване висококалорійне харчування, ферментні препарати для нормалізації функції травного тракту.
За наявності гіпофункції яєчників унаслідок конституціональної недостатності передньої частки гіпофіза, що не асоціюється із затримкою росту, доцільно проводити циклічну вітамінотерапію (комплекс вітамінів групи В та глутамінова кислота по 0,25 г тричі на добу протягом 20 днів; вітамін Е та аскорбінова кислота максимальною дозою до 1 г на день протягом 10 днів), фізіотерапевтичні процедури (ендоназальний електрофорез кальцію, імпульсний струм на білатеральні ділянки, вібромасаж), призначати нейротропні препарати. Важливою складовою лікування є дотримання режиму праці та відпочинку, раціональне харчування.
Терапію проводять під контролем показників базальної температури, результатів кольпоцитології, екскреції 17-КС та естрогенів протягом 3 міс. При контрольному огляді через півроку після закінчення лікування за умови закриття зон росту та відсутності вторинних статевих ознак, пацієнткам віком старше 16 років для стимуляції гіпоталамо-гіпофізарної системи призначають циклічну гормональну терапію естрогенами у низьких дозах, після чого застосовують гестагени.
Для лікування гіпоталамічних форм затримки статевого розвитку використовують препарати гонадоліберину (у пульсуючому режимі по 2 мкг підшкірно або внутрішньом’язово кожні 90 хв з 23.00 год 30 хв до 7.00 год 30 хв три ночі на тиждень протягом 6 тиж).
Крім того, застосовують кломіфену цитрат. Препарат зв’язується з рецепторами 17β-естрадіолу в гіпоталамічних ядрах і, як наслідок, посилюються синтез та секреція гонадоліберинів і тропінів з наступним локальним впливом на біосинтез стероїдів у яєчниках.
За наявності у визначеного контингенту пацієнток дефіциту СТГ застосовують соматотропні та гонадотропні гормони у поєднанні із загальнозміцнюючою терапією, негормональними анаболічними, ферментними препаратами, вітамінами. Лікування проводять спільно з ендокринологом.
За наявності пухлини ЦНС проводиться нейрохірургічна корекція.
Критеріями ефективності лікування є поліпшення показників фізичного та статевого розвитку протягом 3-6 міс і поява самостійних менструацій.
Затримка статевого розвитку яєчникового генезу (дисгенезія гонад)
Частота дисгенезії гонад становить 1 випадок на 10-12 тис. новонароджених, однак це захворювання – значуща соціальна та медична проблема. Причиною патології є природжений дефект статевих хромосом. Перебіг дисгенезії гонад має три основні клінічні форми: типова дисгенезія гонад (синдром Шерешевського – Тернера), чиста і змішана форми дисгенезії гонад.
Синдром Шерешевського – Тернера
Це хромосомна хвороба, що супроводжується характерними аномаліями фізичного розвитку, низькорослістю і статевим інфантилізмом (рис. 5).
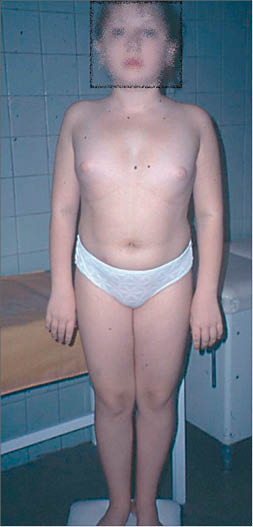 Рис. 5. Пацієнтка із синдромом Шерешевського – Тернера (Баисова Б. И. и др. Гинекология: учебник под ред. Савельевой Г. М., Бреусенко В. Г., 2011)
Рис. 5. Пацієнтка із синдромом Шерешевського – Тернера (Баисова Б. И. и др. Гинекология: учебник под ред. Савельевой Г. М., Бреусенко В. Г., 2011)Синдром Шерешевського – Тернера виникає внаслідок повної або часткової моносомії за Х-хромосомою (45,X0). Під час поділу статевих клітин батьків порушується розходження статевих хромосом, в результаті чого замість нормальної кількості Х-хромосом зародок отримує лише одну.
Уперше цю хворобу як спадкову 1925 р. описав Н. А. Шерешевський, який вважав, що вона зумовлена недорозвиненням статевих залоз та передньої частки гіпофіза і поєднується з природженими вадами внутрішніх органів. У 1938 р. Генрі Тернер виділив характерну тріаду ознак: статевий інфантилізм, шкірні крилоподібні складки на бічних поверхнях шиї та деформацію ліктьових суглобів. Синдром Шерешевського – Тернера – це єдина форма моносомії у живонароджених дівчаток. Слід зазначити, що лише у 50 % пацієнток із даним синдромом спостерігається проста повна моносомія (45,X0); у решти (загалом 30-40 %) хворих – різноманітний мозаїцизм і рідкісні варіанти делецій, ізохромосом, кільцевих хромосом.
Варіанти набору хромосом (каріотип), які виявляють при цитогенетичному дослідженні хворих:
- повна моносомія – 45,X0;
- мозаїчний набір – 45,Х0/46,ХХ; 45,Х0/47, ХХХ та ін.
Частота синдрому становить один випадок на 3000 живонароджених дівчаток.
Слід зазначити, що чіткого зв’язку виникнення цього синдрому з віком і будь-якими захворюваннями батьків не виявлено, проте вагітність зазвичай ускладнюється гестозом, загрозою мимовільного викидня, а пологи часто бувають передчасними і патологічними.
В ембріона первинні статеві клітини закладаються майже в нормальній кількості, однак у другій половині вагітності відбувається їхня швидка інволюція, і до моменту народження дитини кількість фолікулів у яєчнику порівняно з нормою різко зменшена або вони цілком відсутні. Це призводить до вираженої недостатності жіночих статевих гормонів, статевого недорозвинення, у більшості хворих – до первинної аменореї і безплідності. Хромосомні порушення є причиною виникнення вад розвитку. Можливо, що супутні аутосомні мутації також відіграють певну роль у появі вад розвитку, оскільки існують стани, подібні до синдрому Шерешевського – Тернера, але без видимої хромосомної патології та статевого недорозвинення.
При синдромі Шерешевського – Тернера статеві залози зазвичай є недиференційованими сполучнотканинними тяжами, що можуть містити елементи гонад. Рідше зустрічаються рудименти яєчників. Інші патологічні дані відповідають особливостям клінічних проявів. Найбільш важливими змінами кістково-суглобової системи є вкорочення п’ясткових і плеснових кісток, аплазія (відсутність) фаланг пальців, деформація променево-зап’ясткового суглоба, остеопороз хребців. Рентгенологічно при синдромі Шерешевського – Тернера турецьке сідло та кістки склепіння черепа не змінені. При обстеженні внутрішніх органів визначаються вади розвитку серця і великих судин, а також нирок. Виявляються рецесивні гени дальтонізму й інших захворювань.
Порушення кількості чи структури статевих хромосом зумовлює різке недорозвинення тканини яєчників і зниження продукції естрогенів. За відсутності фізіологічного гальмування естрогенами гонадотропної функції гіпофіза відбувається значне підвищення рівня ЛГ і ФСГ у крові хворих на синдром Шерешевського – Тернера. Підвищення рівня гонадотропних гормонів спостерігається вже в 9-11-річному віці, при цьому виділення ФСГ домінує над виділенням ЛГ, що може зумовлювати ранню діагностику цього синдрому.
Доведено, що вже від народження у дітей із синдромом Шерешевського – Тернера помітне відставання у фізичному розвитку. Лише приблизно в 15 % підлітків затримка спостерігається в період статевого дозрівання. Для доношених немовлят характерні мала довжина (42-48 см) і маса тіла до 2500-2800 г. Характерними ознаками синдрому при народженні є надлишок шкіри на шиї та інші вади розвитку, особливо кістково-суглобової та серцево-судинної систем, лімфостаз. Обличчя хворих дуже нагадує обличчя сфінкса: зменшене підборіддя, широке перенісся і гіпертелоризм, епікант, птоз. Для немовляти властиві загальне занепокоєння, порушення смоктального рефлексу, зригування фонтаном, блювання.
Патологічні симптоми синдрому Шерешевського – Тернера наведено в таблиці 3.
Таблиця 3. Клінічні симптоми синдромуШерешевського – Тернера та частота їх виявлення
|
Симптом |
Частота, % |
|
Низький зріст |
100 |
|
Природжена лімфедема |
65 |
|
Крилоподібні складки |
65 |
|
Низький ріст волосся на шиї |
75 |
|
Сплощена грудна клітка |
55 |
|
Коротка шия |
50 |
|
Вальгусне викривлення |
45 |
|
Зміна нігтів на стопах і кистях |
75 |
|
Високе піднебіння |
70 |
Затримка росту прогресує і стає явною у третини хворих до кінця першого року життя, в третини – у 2-5 років, у решти – після 6 років. Збільшення зросту переважно становить 2-3 см на рік, зростові «стрибки» відсутні, кінцевий зріст дорівнює 135-141 см. Маса тіла часто є надмірною. Кістковий вік на кілька років відстає від паспортного, згодом ця невідповідність збільшується.
У частини хворих в ранньому віці відмічається затримка психічного і мовленнєвого розвитку, що свідчить про патологію розвитку нервової системи. У дитини в ранньому дитячому віці характерний зовнішній вигляд:
- низький зріст;
- маленька нижня щелепа;
- відстовбурчені вуха;
- коротка шия з крилоподібними складками;
- низько розташована нижня лінія росту волосся на шиї;
- широка грудна клітка з далеко розставленими сосками;
- втягнені соски;
- наявність епікантуса;
- неправильний ріст зубів;
- опущені кути очей та рота;
- часте викривлення рук у ділянці ліктьових суглобів;
- опуклі нігті на коротких пальцях рук.
У період статевого дозрівання вторинні статеві ознаки не розвиваються. Статеве недорозвинення за наявності синдрому Шерешевського – Тернера вирізняється певною своєрідністю. Найбільш поширеними ознаками є геродермія (патологічна атрофія шкіри, що нагадує старечу), калиткоподібні великі соромітні губи, висока промежина, недорозвинення малих соромітних губ, дівочої перетинки та клітора, лійкоподібний вхід у піхву. Молочні залози у більшості хворих не розвинуті, соски розташовані низько. Вторинне оволосіння з’являється спонтанно і буває мізерним. Матка недорозвинена, статеві залози не розвинуті та представлені сполучною тканиною. Менструація відсутня.
У третини пацієнток наявні вади розвитку інших органів. Часто це вади розвитку серцево-судинної системи (незарощення міжшлуночкової перетинки та великої артеріальної протоки, коарктація аорти, звуження устя аорти), сечовивідних шляхів (недорозвиненість нирок, подвоєння сечоводів, подвоєна і підковоподібна нирка). Синдром Шерешевського – Тернера може супроводжуватися косоокістю, дальтонізмом.
Розумова відсталість у пацієнток із цим синдромом спостерігається рідко, проте дівчатка з каріотипом 45,X0 страждають на просторову дезорієнтацію і тому одержують низькі бали за виконання ігрових тестів і завдань з математики; водночас у вербальних тестах оцінка їх IQ має середні й високі значення. У психічному статусі хворих головну роль відіграє своєрідний інфантилізм з ейфорією в поєднанні з гарною практичною пристосованістю і задовільною соціальною адаптацією.
Слід зазначити, що повнота клінічних проявів, тактика і прогноз залежать від варіанту хромосомного набору. Доведено, що в осіб із синдромом Шерешевського – Тернера виявляється висока (58 %) частота отитів. У 35 % випадків запалення ускладнюється розвитком приглухуватості за провідним типом, що пояснюється аномалією будови середнього вуха. Відмічено схильність до розвитку цукрового діабету (62 %), порушення функції щитоподібної залози. Існує ризик пухлинного переродження недорозвинених гонад, тому щорічно необхідно проводити УЗД органів малого таза. Недорозвиненість внутрішніх статевих органів призводить до вираженого статевого інфантилізму, продукція естрогенів знижується, а секреція гонадотропних гормонів (ФСГ, ЛГ) різко підвищується в пубертатний і постпубертатний періоди. Описані випадки перебігу захворювання зі спонтанною пубертацією і навіть збереженою фертильністю, що, можливо, пов’язано з наявністю здорових клонів клітин у гонадах.
Діагностика
З метою підтвердження діагнозу, окрім визначення характерної клінічної картини, необхідно проводити:
- експрес-метод – дослідження статевого хроматину в клітинах букального епітелію. При мозаїчних варіантах захворювання з наявністю клонів, що містять дві Х-хромосоми, в 11-20 % випадків показники знаходяться в нормі;
- каріотипування. При моносомії виявляють каріотип 45,Х0;
- гормональне дослідження – визначення рівнів естрадіолу, прогестерону, ФСГ, ЛГ, СТГ;
- кольпоцитологію;
- УЗД органів малого таза, органів черевної порожнини і нирок;
- рентгенологічне дослідження кисті з променево-зап’ястковим суглобом.
Диференціальну діагностику синдрому Шерешевського – Тернера необхідно проводити насамперед із синдромом Нунан, при якому фенотипові прояви дуже схожі. Синдром успадковується за аутосомно-домінантним типом. На відміну від синдрому Шерешевського – Тернера при синдромі Нунан відсутня виражена затримка росту. Поширеною є розумова відсталість. Характерна ознака – вкорочення 5-го пальця руки, тоді як для синдрому Шерешевського – Тернера типовим є недорозвинення метакарпальних і метатарзальних кісток. Головна відмінність синдрому Нунан – нормальний вміст Х-хроматину в ядрах слизової оболонки ротової порожнини і наявність нормального жіночого каріотипу 46,XX. При даному синдромі можливі вагітність і народження дитини.
Подібна із синдромом Шерешевського – Тернера клінічна картина спостерігається також у гіпофізарного нанізму. Захворювання має спадковий характер. Найбільш характерною клінічною ознакою є затримка росту і фізичного розвитку хворих. На відміну від синдрому Шерешевського – Тернера зазвичай не спостерігаються соматичні аномалії розвитку. Виявляється знижений рівень СТГ, а вміст статевого хроматину є нормальним (каріотип 46,XX).
Природжена чи набута гіпофункція щитоподібної залози, що виникає внутрішньоутробно або у дітей віком до 3 років, за клінічною картиною подібна до синдрому Шерешевського – Тернера. Проте у цьому випадку відсутні соматичні аномалії, а наявні набряки за типом мікседеми, відмічається ріст волосся на обличчі та тілі (lanugo). Лабораторними методами дослідження виявляється знижений вміст гормонів щитоподібної залози, екскреція гонадотропних гормонів і 17-КС у межах норми, каріотип 46,XX.
Також слід проводити диференціальну діагностику із синдромом Бонневі – Ульріха – аутосомно-домінантною хворобою, при якій у деяких хворих зберігається генеративна функція, спостерігається передача патологічного гена чи генів з покоління в покоління і відсутність характерної цитогенетичної картини моносомії.
Отже, з метою диференціальної діагностики необхідними є визначення відсоткового складу статевого хроматину в епітеліальних клітинах слизової оболонки щоки; каріотипування; УЗД органів малого таза, молочної та щитоподібної залоз; вагіно- та кольпоскопія; визначення рівнів ЛГ та ФСГ, естрогенів, прогестерону, ТТГ, тироксину в плазмі периферійної крові (табл. 4).
Таблиця 4. Диференціально-діагностичні ознаки типової форми дисгенезії гонадта центральної форми затримки росту та статевого розвитку
|
Показники |
Типова формадисгенезії гонад |
Центральна форма затримкиросту та статевого розвитку |
|
Зріст |
Низький |
Низький |
|
Будова тіла |
Диспластична |
Пропорційна |
|
Стигми |
Наявні |
Немає |
|
Вади магістральних судин |
Наявні |
Немає |
|
Кістковий вік |
Відставання від календарного віку |
Відставання від календарного віку |
|
Матка |
У вигляді тяжу |
Невеликого розміру |
|
Гонади |
У вигляді тяжу |
Невеликого розміру |
|
Хромосомні аномалії |
Наявні |
Немає |
|
Рівень гонадотропінів |
Високий |
Низький |
|
Рівень гормона росту |
Норма |
Низький |
|
Проба з гонадоліберинами |
Негативна |
Позитивна |
Лікування
Лікування дітей із синдромом Шерешевського – Тернера має бути комплексним і проводитись у двох напрямках:
- з перших років життя показана терапія, що стимулює ріст (вітаміни, СТГ);
- реконструктивна хірургія природжених вад внутрішніх органів.
У науковій літературі наявні відомості про позитивну динаміку зросту при введенні гормона росту (соматотропін людини, нордитропін, сайзен і т.ін.), за допомогою якого можна досягти збільшення остаточного зросту пацієнтки на 4-8 см.
Анаболічні стероїди (метандростенолон, ретаболіл) рекомендовано призначати низькими дозами і за наявності затримки осифікації (тобто кістковий вік відстає від паспортного більше ніж на 3 роки). Однак це лікування не запобігає розвитку остеопорозу і підвищеному ризику переломів.
Після досягнення дівчатками віку 12-13 років розпочинають терапію жіночими статевими гормонами мінімальними дозами; потім дозу поступово підвищують до рівня, необхідного для розвитку молочних залоз і статевих органів, появи оволосіння. Лікування сприяє фемінізації статури, розвитку жіночих вторинних статевих ознак, насиченню організму статевими гормонами, поліпшенню трофіки статевих органів, знижує підвищену активність гіпоталамо-гіпофізарної системи. Замісну терапію жіночими статевими гормонами слід проводити протягом усього дітородного віку хворих.
Необхідно пам’ятати, що повне вилікування таких пацієнток неможливе. Прогноз при синдромі Шерешевського – Тернера є сприятливим, виняток становлять хворі з тяжкими природженими вадами серця і великих судин та нирковою гіпертензією. Лікування жіночими статевими гормонами сприяє збереженню якості життя пацієнток, однак абсолютна більшість з них залишається безплідною.
Ризик повторного народження дитини з синдромом в родині при нормальному каріотипі батьків не перевищує 1 %.
Чиста форма дисгенезії гонад
Чиста форма дисгенезії гонад (синдром Сваєра, оваріальна дисгенезія) – патологічний стан, який характеризується відсутністю нормально розвинених яєчників. При цитогенетичному дослідженні виявляється збереження обох хромосом, можлива наявність жіночого (46,ХХ), чоловічого (46,ХY) каріотипу та мозаїцизму. Відповідно до каріотипу відсоток статевого хроматину в ядрах епітелію слизової оболонки рота може бути достатнім (14-20 %), низьким або відсутнім. Може спостерігатися світіння Y-хроматину (наявність Y-хромосоми в каріотипі).
Етіологія: генні та хромосомні мутації, загибель первинних статевих клітин (гоноцитів) як наслідок несприятливого впливу на організм матері під час вагітності.
Клінічна картина: відсутність соматичних аномалій розвитку, пропорційна будова тіла, нормальний зріст; індиферентна будова зовнішніх статевих органів, відсутність яєчників (гонадні тяжі), аменорея; відсутність розвитку молочних залоз; затримка дозрівання кісток; підвищення секреції гонадотропінів.
Змішана форма дисгенезії гонад
Для цієї форми характерний каріотип 45Х/46ХY або мозаїцизм з обов’язковою наявністю Y-хромосоми чи її частини, статевий хроматин негативний.
Клінічна картина: відсутність соматичних стигм за наявності ознак вірилізації (різка недорозвиненість малих та великих соромітних губ, гіпертрофія клітора, відсутність молочних залоз, незначно виражений гіпертрихоз). З одного боку на місці гонади визначається сполучнотканинний тяж, з іншого – дисгенетичне яєчко, яке може бути джерелом розвитку злоякісної пухлини (дисгерміноми, адренобластоми).
Лікування усіх форм дисгенезій гонад довготривале. Із пубертатного віку необхідно проводити пожиттєву замісну терапію, спрямовану на забезпечення пропорційного соматичного розвитку, зменшення статевого інфантилізму, підтримання розвитку вторинних статевих ознак, збільшення щільності кісткової маси, відновлення нервово-психічної рівноваги.
При змішаній формі слід видаляти чоловічі гонади для профілактики розвитку пухлин та усунення симптомів вірилізації. З косметичною метою виконують ампутацію клітора, пластичну операцію на шиї (видаляють крилоподібні складки).
Нарушения полового развития у девочек
И. Б. Вовк, В. К. Кондратюк, В. Ф. Петербургская
В статье освещены современные аспекты проблемы нарушения полового созревания у девочек. Представлены клинические проявления основных форм патологии, возможности диагностики и лечения.
Ключевые слова: половое развитие девочек, преждевременное созревание, задержка полового развития.
Disorders of sexual development in girls
I. B. Vovk, V. K. Kondratiuk, V. F. Peterburgskaia
The article highlights the modern aspects of the problem of disorders of sexual development in girls. The clinical manifestations of the main forms of pathology, diagnosis and treatment are presented.
Keywords: sexual development of girls, premature puberty, delayed sexual development.