


27 березня, 2015
Лизосомные болезни накопления
Лизосомные болезни накопления (ЛБН) — это обширный класс наследственных болезней обмена веществ, который включает около 40 нозологических форм. Молекулярные механизмы этиопатогенеза ЛБН сходны. Все они обусловлены генетическими изменениями лизосомных ферментов, контролирующих процесс внутриклеточного расщепления таких макромолекул, как гликозаминогликаны, гликолипиды, гликопротеины. Патогенетическими следствиями этих изменений являются внутрилизосомное накопление нерасщепленных макромолекул и увеличение числа лизосом в клетках различных тканей организма, что морфологически выявляется как наличие так называемых «пенистых» клеток в этих тканях. Такое накопление приводит к нарушению нормального функционирования клеток и их гибели. Чем сильнее функция фермента нарушена мутацией, тем быстрее наступает гибель клеток в тканях и тем быстрее прогрессирует заболевание.

 Накопление нерасщепленных макромолекул при ЛБН может достигать значительных размеров, обусловливая в большинстве случаев несовместимость этих заболеваний с жизнью. Например, при болезни Тея-Сакса вес накопленного ганглиозида достигает 10-15% по отношению к сухой массе головного мозга. Однако известны и обратные примеры, к числу которых принадлежат болезни Краббе и Фабри. Накопление нерасщепленных метаболитов при этих заболеваниях носит умеренный характер и даже не является надежным диагностическим признаком.
Накопление нерасщепленных макромолекул при ЛБН может достигать значительных размеров, обусловливая в большинстве случаев несовместимость этих заболеваний с жизнью. Например, при болезни Тея-Сакса вес накопленного ганглиозида достигает 10-15% по отношению к сухой массе головного мозга. Однако известны и обратные примеры, к числу которых принадлежат болезни Краббе и Фабри. Накопление нерасщепленных метаболитов при этих заболеваниях носит умеренный характер и даже не является надежным диагностическим признаком.
В зависимости от природы накапливаемых макромолекул различают четыре группы ЛБН: мукополисахаридозы, муколипидозы, гликопротеинозы и сфинголипидозы.
Клиническая характеристика, возраст возникновения и тяжесть протекания отдельных заболеваний этих групп варьируют в довольно широких пределах. Они определяются генетическими особенностями нарушений, физиологической значимостью пораженного мутацией метаболического пути, а также тканью-мишенью, в которой нерасщепленные макромолекулы накапливаются.
Так, накопление метаболитов в паренхиматозных органах при некоторых заболеваниях приводит к развитию у больных гепатоспленомегалии с появлением таких признаков гиперспленизма, как анемия и тромбоцитопения (болезнь Гоше, мукополисахаридозы); тогда как ряд болезней протекает без вовлечения печени и селезенки в патологический процесс накопления (метахроматическая лейкодистрофия, болезни Фабри и Краббе).
Накопление метаболитов в костной ткани способствует развитию широкого спектра нарушений, обозначаемых термином «множественный дизостоз». Отмечаются также изменения в суставах, часто с ограничением объема движений в них (мукополисахаридозы, муколипидозы, болезнь Гоше). Хотя некоторые заболевания не имеют признаков поражения костной ткани (метахроматическая лейкодистрофия, болезни Фабри и Краббе).
Накопление нерасщепленных макромолекул в нервной ткани, как правило, обусловливает дегенеративные изменения в центральной нервной системе и развитие умственной отсталости у пациентов (метахроматическая лейкодистрофия, болезнь Краббе, мукополисахаридозы, муколипидозы, гликопротеинозы). Однако некоторые заболевания протекают без вовлечения нервной ткани в патологический процесс накопления и характеризуются нормальным интеллектуальным развитием пациентов (болезни Гоше I типа и Фабри).
Целый ряд заболеваний из групп мукополисахаридозов, муколипидозов и гликопротеинозов отличаются характерным внешним видом пациентов. Большинству таких больных свойственны грубые, гротескные черты лица, с чем связано использование в прошлом названия этих заболеваний «гаргоилизм». Внешний вид пациентов, страдающих другими лизосомными болезнями, такими как болезнь Гоше, метахроматическая лейкодистрофия, болезнь Фабри, не имеет особенностей.
 Таким образом, достаточно четко выражен клинический полиморфизм лизосомных болезней накопления. Однако, несмотря на это, существуют признаки, характерные для всех заболеваний этого класса, а именно:
Таким образом, достаточно четко выражен клинический полиморфизм лизосомных болезней накопления. Однако, несмотря на это, существуют признаки, характерные для всех заболеваний этого класса, а именно:
- полисистемность, то есть вовлечение в патологический процесс многих органов и тканей;
- прогредиентное течение — возникновение и прогрессирование заболевания после некоторого периода нормального развития.
В большинстве своем эти болезни приводят к ранней инвалидизации и преждевременной смерти. Лишь немногие формы болезней характеризуются близкой к норме продолжительностью жизни. Говорят, что такие дети умирают трижды: сначала в умах родителей, когда поставлен диагноз, затем при помещении ребенка в специализированное учреждение, если его туда направляют, и, наконец, когда больной действительно умирает. Безысходность заболевания и серьезные генетические прогнозы формируют сложную психологическую проблему в семье. Отсутствие эффективных способов лечения этих изнуряющих нейродегенеративных заболеваний требует от врача, имеющего дело с родителями больных детей, огромного такта. Трудно передать то опустошающее влияние на семью, которое оказывают быстрое ухудшение состояния и неизбежная гибель ранее здорового ребенка.
Именно поэтому разработка эффективного метода лечения, хотя бы одного заболевания из этой группы фатальных болезней, в высшей степени важна. Первым реальным шагом, предпринятым в этом направлении, стало появление в 1991 году метода лечения болезни Гоше с помощью модифицированной формы недостающего при этом заболевании фермента.
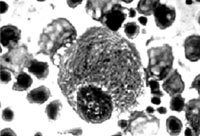
Болезнь Гоше — наследственное заболевание из группы сфинголипидозов, обусловленное недостаточной активностью одного из лизосомных ферментов — глюкоцереброзидазы, которая участвует в гидролизе глюкоцереброзида. В результате, глюкоцереброзид накапливается в лизосомах макрофагов с образованием клеток Гоше, являющихся отличительной особенностью данного заболевания. Этими клетками изобилует красная пульпа селезенки, синусоиды печени, лимфатические узлы, костный мозг, а также многие другие ткани.
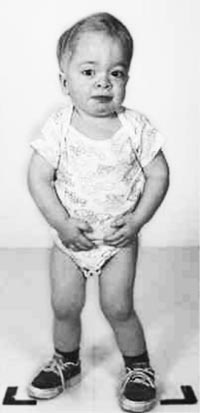
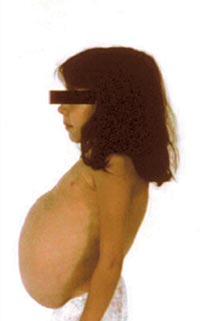
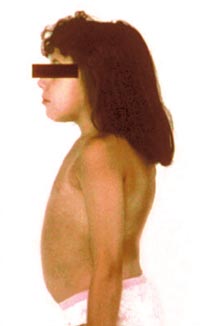
Безболезненная спленомегалия — обычно самый ранний симптом болезни Гоше. Если в норме объем селезенки находится в пределах 50-200 см3, то у пациентов, страдающих болезнью Гоше, он составляет 1500-3000 см3, в отдельных случаях — до 10000 см3 и более. В самых тяжелых случаях вес селезенки может достигать 20% веса тела пациента. У большинства больных гиперспленизм развивается параллельно с панцитопенией и геморрагическим диатезом.
Хотя увеличение печени и ее дисфункция при болезни Гоше являются обычными для всех типов болезни, но случаи тяжелой печеночной недостаточности встречаются редко. Объем печени, как правило, в 1,5-2 раза больше нормы, но в тяжелых случаях может увеличиваться в 10 раз.
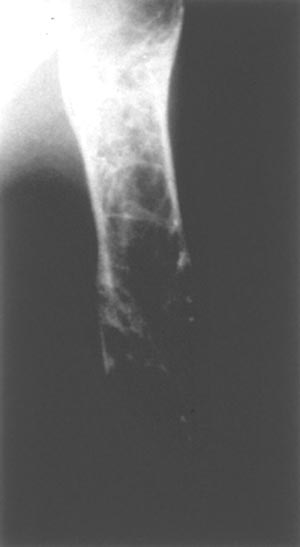 Основной причиной стойкой инвалидизации при этом заболевании является одно из главных осложнений — поражение скелета. Изменения в костной ткани — следствие замещения нормальных элементов костного мозга инфильтратами клеток Гоше, которое сопровождается нарушением нормальных физиологических процессов. Костная патология наблюдается приблизительно у 75% пациентов, страдающих болезнью Гоше. В первую очередь обычно поражается бедренная кость, затем другие трубчатые кости и позвоночник. Накопление клеток Гоше в костном веществе вызывает появление остеолитических очагов, что приводит к отеку, увеличению внутрикостного давления и острым болевым ощущениям, которые известны как «костные кризы».
Основной причиной стойкой инвалидизации при этом заболевании является одно из главных осложнений — поражение скелета. Изменения в костной ткани — следствие замещения нормальных элементов костного мозга инфильтратами клеток Гоше, которое сопровождается нарушением нормальных физиологических процессов. Костная патология наблюдается приблизительно у 75% пациентов, страдающих болезнью Гоше. В первую очередь обычно поражается бедренная кость, затем другие трубчатые кости и позвоночник. Накопление клеток Гоше в костном веществе вызывает появление остеолитических очагов, что приводит к отеку, увеличению внутрикостного давления и острым болевым ощущениям, которые известны как «костные кризы».
Следует отметить, что болезнь Гоше представляет собой группу из трех клинически различных, но генетически общих нозологических форм. Кроме вышеприведенных симптомов, характерных, как правило, для всех типов болезни Гоше, существует и принципиальное различие между типами, которое состоит в наличии и скорости прогрессирования неврологических осложнений. Так, первый, самый распространенный тип этого заболевания не затрагивает нервной системы; на первый план выступает увеличение паренхиматозных органов с преимущественным поражением селезенки. Второй и третий типы характеризуются развитием неврологических осложнений у пациентов в разном возрасте.
Таким образом, в случае неясной спленомегалии (с геморрагическим диатезом и без него) у пациента любого возраста, особенно в случаях безболезненного увеличения селезенки в сочетании с другими патологическими проявлениями со стороны печени и скелета, всегда нужно помнить о вероятности наличия у больного болезни Гоше.
Наличие клеток Гоше в костном мозге и других тканях с высокой степенью вероятности подтверждает диагноз болезни Гоше. Однако он не может быть принят как основополагающий, так как подобные клетки встречаются и при других лизосомных болезнях (болезнь Нимана-Пика и другие). Поэтому для подтверждения диагноза, наряду с гистологическим исследованием костного мозга, необходимо определять активность мутантного фермента в клетках периферической крови.
Программа диагностики наследственных лизосомных болезней накопления, включая болезнь Гоше, существует в лаборатории Киевского медико-генетического центра Украинской детской специализированной больницы ОХМАТДЕТ. В тесном сотрудничестве с Центром детской онкогематологии УДСБ ОХМАТДЕТ региональные медико-генетические центры и другие медицинские учреждения Украины проводят комплексное клинико-лабораторное обследование пациентов с этой патологией.
До недавнего времени медицина предлагала только симптоматические методы лечения болезни Гоше. Единственным способом облегчить такие тяжелые проявления гиперспленизма, как анемия и тромбоцитопения, было удаление селезенки. Проведение спленэктомии у этих пациентов позволяло достичь временного облегчения. Однако накопление нерасщепленного глюкоцереброзида в организме не прекращалось, и основная нагрузка при отсутствии селезенки ложилась на костную ткань. Таким образом, эта операция приводила к ускорению деструктивных изменений в костной системе.
С выделением и очисткой глюкоцереброзидазы стало возможным замещение мутантного фермента у больных. Десятилетний опыт применения ферментозаместительной терапии во всем мире свидетельствует о том, что этот метод лечения останавливает прогрессирование заболевания, способствует обратному развитию симптомов болезни Гоше и значительно улучшает качество жизни больных. Сегодня во всем мире тысячи больных с болезнью Гоше получают внутривенные инъекции модифицированной человеческой глюкоцереброзидазы. В Украине двум детям проводится специфическое лечение в качестве гуманитарной помощи фирмы Джензайм.
 На сегодняшний день болезнь Гоше занимает среди лизосомных болезней накопления особое положение модельной системы, в соответствии с которой должно развиваться исследование всех других нозологических форм такого обширного класса. Для этой патологии установлен первичный биохимический дефект, исследована структура нормального белка и нормального гена, разработан и внедрен в практику метод ферментозаместительной терапии. В настоящее время за рубежом получают специфическое лечение больные с болезнями Фабри, мукополисахаридозом I и II типов, в стадии клинических испытаний находятся аналогичные методы коррекции болезни Помпе, ведутся исследования по возможности использования для этой цели генной терапии.
На сегодняшний день болезнь Гоше занимает среди лизосомных болезней накопления особое положение модельной системы, в соответствии с которой должно развиваться исследование всех других нозологических форм такого обширного класса. Для этой патологии установлен первичный биохимический дефект, исследована структура нормального белка и нормального гена, разработан и внедрен в практику метод ферментозаместительной терапии. В настоящее время за рубежом получают специфическое лечение больные с болезнями Фабри, мукополисахаридозом I и II типов, в стадии клинических испытаний находятся аналогичные методы коррекции болезни Помпе, ведутся исследования по возможности использования для этой цели генной терапии.
Благодаря успехам генетики и современной медицинской науки эффективное лечение наследственных болезней становится реальностью, уменьшает боль и страдания многих людей, дарит ранее безнадежным пациентам надежду на полноценную жизнь.