


4 травня, 2017
Морфогенетические белки FGF23 и Klotho, гиперфосфатемия и карбамилирование белков – нетрадиционные факторы риска кардиоваскулярных событий у больных хронической болезнью почек

 Количество больных хронической болезнью почек (ХБП) во всем мире составляет более 497,5 млн человек, и основной причиной терминальной стадии ХБП в большинстве развитых стран в настоящее время является сахарный диабет (СД) [15]. Его распространенность в мире достигла эпидемических размеров, к 2040 году предполагается увеличение количества таких больных до 642 млн человек [6]. Согласно данным Американской диабетической ассоциации у 40% больных СД развивается диабетическая нефропатия (ДН) [1], которая у значительного количества этих больных будет прогрессировать до терминальной стадии ХБП [11], однако пациенты с ХБП чаще умирают от сердечно-сосудистых причин, чем от почечной недостаточности [14]. Риск смерти особенно высок при болезнях почек поздних стадий; 30-летний пациент с терминальной стадией почечной недостаточности оказывается перед эквивалентным риском смерти 90-летнего человека без ХБП [22]. Помимо медицинских аспектов ХБП – существенная экономическая нагрузка на систему здравоохранения даже для стран с высокими доходами, что вынуждает усиливать научные исследования, направленные на улучшения прогноза и качества жизни людей с диабетом и болезнью почек.
Количество больных хронической болезнью почек (ХБП) во всем мире составляет более 497,5 млн человек, и основной причиной терминальной стадии ХБП в большинстве развитых стран в настоящее время является сахарный диабет (СД) [15]. Его распространенность в мире достигла эпидемических размеров, к 2040 году предполагается увеличение количества таких больных до 642 млн человек [6]. Согласно данным Американской диабетической ассоциации у 40% больных СД развивается диабетическая нефропатия (ДН) [1], которая у значительного количества этих больных будет прогрессировать до терминальной стадии ХБП [11], однако пациенты с ХБП чаще умирают от сердечно-сосудистых причин, чем от почечной недостаточности [14]. Риск смерти особенно высок при болезнях почек поздних стадий; 30-летний пациент с терминальной стадией почечной недостаточности оказывается перед эквивалентным риском смерти 90-летнего человека без ХБП [22]. Помимо медицинских аспектов ХБП – существенная экономическая нагрузка на систему здравоохранения даже для стран с высокими доходами, что вынуждает усиливать научные исследования, направленные на улучшения прогноза и качества жизни людей с диабетом и болезнью почек.
Традиционные факторы риска, такие как курение табака, артериальная гипертензия, диабет и гиперхолестеринемия, являются главными определяющими факторами кардиоваскулярных событий в общей популяции. На индивидуальном уровне, однако, подверженность к развитию атеросклероза изменяется значительно, но роль других заболеваний в его патогенезе изучена недостаточно. В клинической практике очень часто встречаются коморбидные состояния с развитием острой почечной недостаточности или ХБП, однако общепринятые факторы сердечно-сосудистого риска не объясняют высокий уровень смертности от кардиоваскулярных событий при таком сочетании.
Одними из чрезвычайно важных составляющих патогенеза осложнений болезней почек являются нарушения минерального обмена. В 2006 году, после того как было установлено, что минеральные и скелетные нарушения, сопутствующие почечной недостаточности, являются важными компонентами сердечно-сосудистых осложнений у больных ХБП и высокой летальности, был принят термин CKD-MBD [16]. Синдром CKD-MBD предполагает биохимические изменения минерального обмена, нарушение скелетного ремоделирования и внескелетный кальциноз, которые развиваются при снижении гломерулярной фильтрации более чем на 40%. При определении концепции синдрома CKD-MBD, связанной с изучением сердечно-сосудистых факторов риска, учитывают три новых признака:
1) изменения содержание фосфора [3];
2) отклонения в содержании фактора роста фибробластов 23 (FGF23) [7];
3) наличие сосудистого кальциноза [10].
Отложение кальция в сосудах представляет собой особенно высокую угрозу летальности от сердечно-сосудистых осложнений и смертности от всех причин [17]. Патологический кальциноз мягких тканей оказывает вредные воздействия в зависимости от включаемой органной системы. Нефрокальциноз – депонирование кальция в пределах почечной паренхимы и канальцев. Это накопление кальция в почке обусловлено увеличением экскреции с мочой кальция, фосфора и/или оксалатов в комбинации с потерей защитных ингибиторов минерализации в моче. В зависимости от этиологии нефрокальциноз может вызвать прогрессирующую почечную дисфункцию и терминальную стадию почечной недостаточности [12]. Подобно нефрокальцинозу сосудистый кальциноз вызван дисбалансом прокальцифицирующих и противокальцифицирующих факторов. Принимая во внимание, что кальциноз рассматривается как компонент старения, увеличенный кардиоваскулярный риск особенно распространен при таких заболеваниях, как СД и ХБП.
Дополнительное влияние на развитие кардиоваскулярных событий при ХБП оказывают нарушения гомеостаза, обусловленные изменением метаболизма как липидов, так и белковых субстратов с развитием карбамилирования при нарушении азотовыделительной функции почек [5]. Особо необходимо выделить изменения минерального обмена, такие как гиперфосфатемия, увеличение содержания паратгормона (PTH), FGF23 и недостатка витамина D [20]. FGF23 является циркулирующим гормоном, который продуцируется остеоцитами и центральная роль которого заключается в контроле минерального обмена [18]. FGF23 снижает уровень фосфора в сыворотке и косвенно понижает уровень содержания кальция в сыворотке путем его ингибирующего воздействия на концентрацию витамина D и PTH [7-10]. Предполагается, что FGF23 также непосредственно принимает участие в развитии патологических процессов в сосудистой сети [11] и сердце [12]. Увеличенное содержание фосфора и 1,25-дигидроксивитамин D (1,25 (OH)2D) стимулируют выделение FGF23 из остеоцитов под контролем PHEX и DMP1 белков [13]. Интактный FGF23 связывается с его корецептором α-Klotho, который секретируется почечными канальцевыми клетками. Это комплексное соединение активирует рецепторы FGF в почечных проксимальных канальцах и уменьшает экспрессию II типа натрий-фосфатного котранспортера (NaPi2) [17]. Таким образом FGF23 предотвращает гиперфосфатемию, уменьшая повторную абсорбцию фосфатов из мочи. Этот эффект заметно выражен при ХБП, когда уровень FGF23 увеличивается, чтобы противостоять уменьшенной гломерулярной фильтрации фосфора при снижении скорости гломерулярной фильтрации [8]. FGF23 также ингибирует CYP27B1 (25-гидроксивитамин D‑1-α-гидроксилазу), снижающую продукцию 1,25 (OH)2D, и стимулирует CYP24A1 (1,25 дигидрооксивитамин D‑24-гидроксилазу), увеличение конверсии 1,25 (OH)2D в неактивные метаболиты. В исследовании Тhe Ludwigshafen Risk and Cardiovascular Health Study (LURIC) изучали, была ли концентрация c-терминала FGF23 связана с сердечно-сосудистой и общей летальностью у 2974 пациентов, у которых была проведена коронарография [20]. Средний возраст участников составил 63±10 лет; средние уровни с-терминала FGF23 в сыворотке – 54 (40-78) RU/мл. Наблюдая за пациентами в течение 9,9 года, авторы установили, что 884 участника умерли (30%), среди них 545 (18%) – вследствие сердечно-сосудистых причин. При этом FGF23 значительно и инверсно коррелировал с уровнем скорости клубочковой фильтрации (СКФ). Таким образом, почти у 3 тыс. пациентов, подвергшихся коронарографии и 9,9 года наблюдения, содержание с-терминала FGF23 до лечения предсказывало риск смерти от всех причин и сердечно-сосудистой летальности. Анализ показал линейную зависимость между FGF23 и исходами болезни. У участников исследования с комбинацией высокого FGF23 (>60 пг/мл), низкой СКФ (<60 мл/мин), микро- или макроальбуминурией и соотношением альбумин/креатинин выше 3 мг/мл зафиксировано почти восьмикратное увеличение риска смерти по сравнению с участниками без данных расстройств. Эта связь была независима от стандартных сердечно-сосудистых факторов риска и уровня фосфатов в сыворотке крови.
Вместе с тем повышение уровня фосфатов в сыворотке крови было связано с увеличенным сердечно-сосудистым риском даже при популяционных исследованиях, а незначительные изменения FGF23, в пределах нормальной амплитуды у здоровых индивидуумов, являются независимыми факторами риска сердечно-сосудистых событий [4]. Это может быть обусловлено и тем, что при гиперфосфатемии увеличивается уровень FGF23. Гиперфосфатемия, в свою очередь, может развиваться из-за увеличения содержания фосфора в продуктах питания в связи с использованием фосфатных добавок, которые являются главным образом неорганическими солями с быстрым всасыванием в кишечнике и высокой биодоступностью [2].
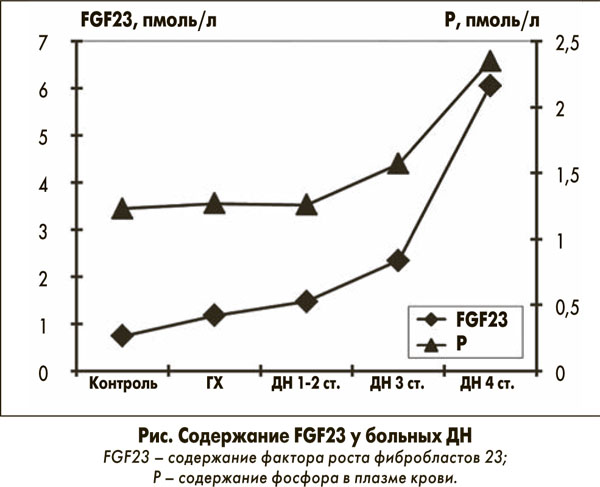
Нарушениям структуры сосудистой стенки предшествует дисфункция эндотелия и предполагается, что уже на начальных стадиях ХБП нарушения фосфорно-кальциевого обмена играют важную патогенетическую роль в дальнейшем развитии кальциноза и тромбоза артериальных сосудов. По нашим данным, содержание FGF23 у больных ДН прямо коррелирует с уровнем гиперфосфатемии (рис.), нарушением функционального резерва почек и случаев развития ишемической болезни сердца у больных ДН [2], что согласуется с результатами C.M. Rebholz и соавт., 2015 и T. Liyanage и соавт., 2015 [11, 18].
Изменения фосфорно-кальциевого обмена способствуют также развитию гипертрофии левого желудочка (ГЛЖ). При проведении корреляционного анализа между состоянием миокарда и активностью системы FGF23 / фосфорно-кальциевый обмен у 260 больных ДН нами установлена положительная корреляционная связь между уровнем фосфора и ГЛЖ (r = +0,48; р<0,05). Чрезвычайно высокая прямая корреляционная связь отмечена на поздних стадиях ДН, особенно при наличии почечной недостаточности, и установлена связь между концентрацией фосфора и объемными показателями ЛЖ (конечным диастолическим объемом, КДО и конечным систолическим объемом, КСО) (r = +0,68; р<0,01), а устойчивая обратная корреляционная связь – с фракцией выброса ЛЖ (r = -0,56; р<0,01). Установлена прямая корреляционная связь между уровнем FGF23 и индексом массы ЛЖ, особенно на более поздних стадиях ДН, начиная с третьей стадии (r = +0,58; р<0,05), что указывает на изменение данного показателя при формировании соединительно-тканного каркаса миокарда. Выявлена устойчивая прямая корреляционная связь между уровнями FGF23 и показателями ГЛЖ: толщиной задней стенки ЛЖ и межжелудочковой перегородки (от r = +0,48 до r = +0,56, в среднем r = +0,53; p<0,05), а также между изменением концентраций FGF23 и объемными показателями ЛЖ (КДО и КСО) (r = +0,64; р<0,01). На поздних стадиях ДН при наличии сердечной недостаточности установлена обратная корреляционная связь между концентрацией FGF23 и фракцией выброса ЛЖ. Наши данные о FGF23 согласуются с результатами, полученными в исследовании CRIC, где повышение уровня FGF23 определялось уже на второй стадии ДН.
Точные пути воздействия FGF23 на сердечно-сосудистую систему еще недостаточно изучены. Известно, что медиатором для FGF23 в сосудистой стенке является Klotho – эволюционно сохранившийся белок, связанный с продолжительностью жизни. Ген и белок α-Klotho были обнаружены в 1997 году Kuro-о и сотрудниками. Название Klotho связано с греческой мифологией, где Klotho – одна из судеб, которая прядет нить жизни. Белок Klotho у млекопитающих присутствует в различных изоформах – как мембраносвязанный белок и как растворимая форма. Растворимый Klotho может генерироваться, теряя внеклеточный домен мембранного Klotho, а мембранный Klotho способен формировать комплексное соединение с рецепторами фактора роста фибробластов и, таким образом, создавать связывающие сайты для FGF23. Нарушение азотовыделительной функции почек является потенциальным состоянием дефицита Klotho, обусловленного снижением его концентрации и в ткани, и в системе кровообращения. Большое значение имеет уточнение, способствует ли недостаток Klotho уменьшению продолжительности жизни и другим многим тяжелым осложнениям у больных с ХБП с увеличением сердечно-сосудистых осложнений и смерти. В различных моделях растворимый Klotho продемонстрировал протективные свойства при остром повреждении почек, почечном фиброзе, уремической кардиомиопатии, сосудистом кальцинозе и эндотелиальной дисфункции. Эти наблюдения очень важны не только для того, чтобы понять патогенетические механизмы заболеваний, но и для разработки более эффективного лечения и профилактических мер, включая замену потерянного гормона.
Экспериментальные исследования показали, что растворимый Klotho как гуморальный фактор может предохранять от повреждения сосудистую сеть. Доставка гена Klotho аденовирусным вектором увеличила эндотелий-зависимый синтез оксида азота и предотвратила неблагоприятное сосудистое ремоделирование в склерозированных артериях страдающих ожирением крыс. Недавно было показано, что растворимый Klotho регулирует сосудистый тонус. In vitro было подтверждено его стимулирующее действие на продукцию оксида азота [3]. Растворимый Klotho подавлял экспрессию адгезивной молекулы сосудистой клетки‑1 и межклеточной молекулы адгезии‑1 в эндотелиальных клетках in vitro, а также ингибировал фосфат-индуцированый кальциноз гладкомышечных клеток. Экспрессируют ли сосудистые гладкомышечные клетки эндогенный Klotho – в настоящее время является предметом изучения. Однако недавно был определен рецептор фактора роста фибробластов, опосредующий влияние FGF23 на развитие ГЛЖ. В декабре 2011 года C. Faul и соавт. идентифицировали рецептор FGF4 (FGFR4) как изоформу, которая опосредует влияние FGF23 на сердечные миоциты [6]. Введение FGF23 индуцировало гипертрофию миоцитов мышей in vitro, но этот эффект блокировался ингибитором FGFR4. Поскольку предшествующие исследования показали, что полное ингибирование активности FGF23 эффективно при редуцировании ГЛЖ [13], эти новые наблюдения предполагают, что FGFR4 или его сигнальные пути могли бы быть терапевтическими целями при попытках уменьшить сердечно-сосудистые осложнения у больных ХБП. Специфические ингибиторы FGFR4 в настоящее время находятся в стадии клинических испытаний и в ближайшие годы могут стать одним из эффективных препаратов лечения нарушений структуры сердца и гемодинамики в целом. Это очень интересные новые находки при изучении осложнений ХБП, однако, как большинство других исследований в этом поле, они ограничены в связи с недостаточностью клинических данных.
Поскольку высокую смертность от сердечно-сосудистых осложений у пациентов с ХБП нельзя объяснить только традиционными факторами риска развития сердечно-сосудистых заболеваний (ССЗ), было высказано предположение, что уремия, при которой в результате снижения СКФ накапливаются токсические продукты, в том числе мочевина, вносит вклад в увеличение риска развития ССЗ при ХБП. Установлено, что высокие уровни мочевины способствуют изменениям белков или аминокислот за счет процесса, называемого карбамилированием – посттрансляционного изменения белков, происходящего при ряде патологических состояний, в том числе молекулярном старении, воспалении и нарушении функции почек, например, при хронической и терминальной почечной недостаточности. Мочевина как продукт белкового катаболизма спонтанно разлагается в водных растворах, формируя циановую кислоту и ее основание, сопряженное с кислотой – цианат [5]. Циановая кислота находится в равновесии с его химически активной формой – изоциановой кислотой. Плазменная концентрация изоциановой кислоты у здоровых людей составляет ~50 нмоль/л, однако может достигать 150 нмоль/л у больных ХБП [8]. Кроме того, цианат может также генерироваться через ферментное каталитическое окисление псевдогалоидного соединения (SCN–) миелопероксидазой (MPO) [15]. MPO – составная часть белков в лейкоцитах (нейтрофилах и моноцитах), значительное количество которых в каталитически активной форме обнаруживается в участках атеросклеротических поражений [23] и нестабильных бляшках у больных ишемической болезнью сердца [19]. Исследования с нокаутной MPO и МРО трансгенных мышей подтверждают, что MPO катализирует карбамилирование белков in vivo [21]. Кроме того, карбамилирование гемоглобина нарушает транспорт кислорода, при этом плазменный уровень гомоцитруллина (PBHCit), получающегося при карбамилировании, является предиктором серьезных неблагоприятных кардиальных событий у пациентов даже с нормальной функцией почек, однако детали такой взаимосвязи при различных стадиях ХБП еще мало изучены [26]. Будучи связанным с уремией, воспалением и сосудистыми заболеваниями, карбамилирование липопротеинов представляет особый интерес при расшифровке риска ССЗ, необъяснимого наличием традиционных факторов риска. Недавние исследования показали, что происходящее в условиях in vivo карбамилирование липопротеинов способствует развитию проатерогенных биологических эффектов, например дисфункции эндотелия и клеточной смерти, образованию пенистых клеток и пролиферации гладкомышечных клеток сосудов.
Карбамилированные белки вызывают особенный интерес как биомаркеры, поскольку их уровень может количественно отражать тяжесть патологического состояния (воспаления и уремии), и они присутствуют в плазме или цельной крови. Помимо белков карбамилированию подвергается и гемоглобин в результате ковалентного связывания изоциановой кислоты с N-концевым остатком глобина. Таким образом, карбамилирование липопротеинов (кЛПНП) приводит к развитию атеросклероза путем сложных биохимических реакций, при этом нарушаются различные защитные механизмы. Поскольку у кЛПНП снижено сродство к печеночным рецепторам ЛПНП, это приводит к уменьшению их клиренса из системы кровообращения. Вместе с тем у кЛПНП отмечается повышенное сродство к макрофагальным скавенджер-рецепторам, что приводит к накоплению холестерола в моноцитах и формированию пенистых клеток. Инкубация кЛПНП с эндотелиальными клетками усиливает адгезию к ним моноцитов, приводит к ускоренному их апоптозу, способствует макрофагальному воспалению и пролиферации гладкомышечных сосудов. По нашим данным, апоптоз моноцитов у больных ХБП значительно повышается и коррелирует с количеством апоптозных, слущенных клеток эндотелия. Помимо структурных изменений сосудистой стенки кЛПНП стимулируют лектиноподобные рецепторы липопротеинов малой плотности 1 (LOX‑1) в эндотелии сосудов, вызывая индуцированную NADPH-окидазой продукцию кислородных радикалов (ROS) и разобщение эндотелиальной синтазы оксида азота (eNOS) [9]. Напротив, карбамилирование липопротеинов высокой плотности (ЛПВП) является одним из механизмом потери их противосклеротической и антиапоптозной активности [5]. Вместе с тем механизм влияния гиперфосфатемии на развитие атеросклероза и ишемической болезни сердца требует дальнейших исследований. Основой лечения гиперфосфатемии в настоящее время являются фосфатбиндеры – проведенные раннее наблюдательные исследования показали снижение риска ССЗ при коррекции содержания фосфатов у больных ХБП, что позволило T.J. Ellam еще в 2012 году предположить: «поскольку атерогенная роль фосфатов продемонстрирована, фосфатбиндеры могли бы стать новыми статинами». В этой связи основу профилактики кардиоваскулярных осложнений у пациентов с ХБП составляют коррекция липидного обмена и попытки корректировать минеральный гомеостаз с использованием оральных фосфатбиндеров, активированного витамина D или аналогов витамина D и кальцимиметиков. Однако их эффективность является недостаточно высокой, и в настоящее время проводятся международные многоцентровые исследования по изучению влияния ряда препаратов на изменения фосфорно-кальциевого обмена. Так, в исследовании COMBINE сейчас изучается влияние комбинированной терапии лантанума карбоната и никотинамида на фосфорный обмен. О результатах данного исследования, как ожидается, будет сообщено в конце 2018 года. В конце 2015 года в США было инициировано исследование III фазы по применению тенапанора для лечения гиперфосфатемии у пациентов ESRD на диализе (Phased 3 registration trials of tenapanor for the treatment of hyperphosphatemia for ESRD patients on dialysis). Тенапанор – мощный ингибитор Na+/H+ транспортера 3 (NHE3), локализованного в апикальной мембране кишечных эпителиальных клеток.
В настоящее время изучаются также стратегии, направленные на предотвращение карбамилирования белков как новейшие подходы к лечению и профилактике атеросклероза. Ряд исследований показали, что терапевтические подходы, направленные на перехват или предотвращение белкового карбамилирования in vivo, могут быть новым фармакологическим подходом для профилактики и лечения сердечно-сосудистых осложнений, особенно среди субъектов с ХБП или терминальной стадией почечной недостаточности [15, 17, 18]. Недавно проведенное исследование, охватившее 23 пациента на гемодиализе, показало перспективный потенциал терапии аминокислотами для уменьшения карбамилирования у больных ХБП [19]. Исследуется возможность профилактики карбамилирования наборами аминокислот – в настоящее время проводится многоцентровое исследование CarRAAT-2, результаты которого ожидаются в 2018 году.
Выводы
1. Гиперфосфатемия может быть прогнозирующим фактором развития сердечно-сосудистых осложнений при ХБП.
2. FGF23 является независимым предиктором прогрессирования ГЛЖ и развития сердечной недостаточности у больных ДН.
3. Доказательство защитного эффекта белка Klotho на сосуды в клинике может играть важную роль в лечении и профилактике кардиоваскулярных осложнений у больных ХБП.
4. Разрабатываемые в настоящее время препараты для лечения гиперфосфатемии на основе ингибиторов NHE3 и блокаторы FGFR4 могут стать более эффективным средством, чем применяемые в клинической практике фосфатбиндеры.
5. Эффективность предупреждения карбамилирования с помощью аминокислот будет установлена после окончания многоцентрового исследования CarRAAT‑2.
Литература
1. American Diabetes Association. Standards of medical care in diabetes‑2014 // Diabetes Care. – 2014. – V. 37. – Suppl 1. – P. 14-80.
2. Calvo M.S., Moshfegh A.J., Tucker K.L. Assessing the health impact of phosphorus in the food supply: issues and considerations //Adv Nutr. 2014; 5: 104-113.
3. Chang J.R., Guo J., Wang Y. et al. Intermedin1-53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of α-Klotho // Kidney Int. 2016. – V. 89. – P. 586-600.
4. di Giuseppe R., Kuhn T., Hirche F., Buijsse B. Fibroblast growth factor 23, its correlates and risk of myocardial infarction: Results from the EPIC-Germany case-cohort study // Atherosclerosis. – 2015. – V. 241. – Issue 1. – Pages e20.
5. Frederik H. Verbrugge, Wilson W.H. Tang and Stanley L Hazen. Protein carbamylation and cardiovascular disease // Kidney Int. – 2015. – V. 88. – P. 474-478.
6. Faul C. FGF23 induces left ventricular hypertrophy / Faul C., Amaral A.P., Oskouei B. et al. //J Clin Invest. – 2011. – Vol. 121. – P. 4393-4408.
7. International Diabetes Federation // IDF Diabetes Atlas. 7th Edition, 2015.
8. Isakova T., Sprague S.M. Rationale and Approaches to Phosphate and Fibroblast Growth Factor 23 Reduction in CKD // J Am Soc Nephrol. – 2015. Oct. – V. 26 (10). – P. 2328-2339.
9. Joseph M. Rutkowski, Johanne Pastor, Kai Sun Adiponectin alters renal calcium and phosphate excretion through regulation of Klotho expression // Kidney Int. – 2017. – V. 91. – Is. – P. 324-337.
10. Lim K. Vascular klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor // Circulation. – 2012. – Vok. 125. – P. 2243-2255.
11. Liyanage T., Ninomiya T., Jha V. et al. Worldwide access to treatment for end-stage kidney disease: a systematic review // Lancet. – 2015. – V. 385. – P. 1975-1982.
12. Marta Zielinska, Andrzej Wasilewski, Jakub Fichna Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome // Expert Opinion on Investigational Drugs. – 2015. – V. 24. – I. 8. – P. 1093-1099.
13. Martin A., Liu S., David V. et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling // FASEB J. 2011; 25: 2551-2562.
14. Mendoza J.M., Isakova T., Cai X., Bayes L.Y., Faul C., Scialla J. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease // Kidney Int. – 2017. – V. 91. – I.3. – P. 711-719.
15. Mills K.T., Xu Y., Zhang W. et al. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010 // Kidney Int. – 2015. – V. 88. – P. 950-957.
16. Moe S. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) / Moe S., Drueke T., Cunningham J. et al. // Kidney Int. – 2006. – Vol. 69. – P. 945-1953.
17. Nordholm A.A potential kidney – bone axis involved in the rapid minute-to-minute regulation of plasma Ca(2+) / Nordholm A., Mace M.L., Gravesen E., Olgaard K., Lewin E. // BMC Nephrol. – 2015. – Vo. 16. – P. 29.
18. Rebholz C.M., Grams M.E., Coresh J. et al. Serum fibroblast growth factor‑23 is associated with incident kidney disease // J Am Soc Nephrol. – 2015. – V. 26. – P. 192-200.
19. Ritter C.S., Slatopolsky Е. Phosphate Toxicity in CKD: The Killer among Us // CJASN. – 2016. – V. 11 (6). – P. 1088-1100.
20. Vincent M. Brandenburg Marcus E. Kleber, Marc G. Vervloet, Andreas Tomaschitz, Stefan Pilz. Fibroblast growth factor 23 (FGF23) and mortality: The Ludwigshafen Risk and Cardiovascular Health Study// Atherosclerosis. – 2014. – V. 237. – Iue 1. – P. 53-59.
21. V. Jha, M. Arici, Allan J. Collins. Understanding kidney care needs and implementation strategies in low- and middle-income countries: conclusions from a «Kidney Disease: Improving Global Outcomes» (KDIGO) Controversies Conference // Kidney Int. – 2016. – V. 90. – Is. 6. – P. 1164-1174.
22. Umut Selamet, Hocine Tighiouart, Mark J. Sarnak Relationship of dietary phosphate intake with risk of end-stage renal disease and mortality in chronic kidney disease stages 3-5: The Modification of Diet in Renal Disease Study // Kidney Int. – 2016. – V. 89. – Is. 1. – P. 176-184.
23. Yamada S., Giachelli C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23 and Klotho // Bone. – 2016. – Nov 12. – P. 8756-3282.