


19 травня, 2017
Взаимозаменяемость оригинальных аналогов инсулина и их биосимиляров: нормативная база, дизайн исследований и клинические последствия

Применение биосимиляров регулируется несколько иначе, чем использование низкомолекулярных генерических лекарственных средств. Обусловлено это тем, что из-за сложного строения биологических препаратов даже небольшие отклонения в технологии производства или составе могут приводить к значимым изменениям эффективности и безопасности. Поэтому для подтверждения взаимозаменяемости биосимиляров с оригинальными препаратами необходимы более строгие и специфичные исследования. В данной статье представлен обзор существующей нормативной базы по подтверждению взаимозаменяемости биосимиляров с особым акцентом на аналогах инсулина.
Поскольку срок патентной защиты ряда сложных биологических препаратов истек или истекает, появилась возможность выхода на фармацевтический рынок биосимиляров, которые могли бы уменьшить стоимость биологической терапии и тем самым повысить ее доступность. С учетом наличия большого и постоянно растущего рынка для таких биологических препаратов, как инсулин, биосимиляры становятся все более привлекательными объектами для многих фармацевтических компаний.
Однако следует отметить, что на пути к утверждению биосимиляров имеются более существенные регуляторные барьеры по сравнению с регистрацией генерических низкомолекулярных препаратов. От производителя биосимиляра требуется предоставить значительно больше данных из-за сложности строения биологических/биотехнологических лекарственных средств, их потенциальной иммуногенности, а также возможного влияния на эффективность и безопасность технологии производства и лекарственной формы (например, готовый к употреблению раствор в картридже или шприц-ручке, концентрированный раствор или порошок во флаконе для разведения).
Программа разработки каждого биосимиляра оценивается в индивидуальном порядке, начиная с используемой клеточной линии, всех этапов производства (ферментация, очистка) и контроля качества, даже если действующее вещество является подобным оригинальному, и заканчивая рядом доклинических (с акцентом на in vitro) и клинических исследований (только фазы I и III).
Но даже в том случае, когда в соответствии со стандартами регуляторных органов были продемонстрированы эффективность и безопасность биосимиляра, остается открытым вопрос о достаточности представленных данных для доказательства его взаимозаменяемости с референтным продуктом. Взаимозаменяемостью называют возможность замены одного препарата на другой, который по ожиданиям должен обеспечить аналогичный клинический эффект в определенной клинической ситуации у любого отдельно взятого пациента.
С биосимилярами аналогов инсулина связаны особые трудности из-за сложности производственного процесса, причем даже несущественные различия в активности или наличии примесей могут стать причиной изменения клинических эффектов. Инсулин характеризуется узким терапевтическим окном и требует очень точного дозирования для сохранения баланса между эффективностью и безопасностью. Кроме того, эффективность, переносимость и приверженность пациента к инсулинотерапии в значительной степени зависят от применяемого доставочного устройства, например шприц-ручки или инсулинового шприца. Удобство и надежность таких устройств является дополнительным вызовом для производителей, хотя и не является предметом обсуждения в данной статье. Но с учетом всего этого демонстрация взаимозаменяемости биосимиляров аналогов инсулина, вероятно, будет более сложной задачей и потребует специально спланированных клинических исследований в дополнение к тем, которые необходимы для подтверждения биосимилярности.
Некоторые производители неинсулиновых биосимиляров уже провели отдельные перекрестные исследования (с переводом пациентов с одного препарата на другой), чтобы подтвердить взаимозаменяемость с референтным лекарственным средством. Следует также отметить, что практика проведения перекрестных исследований уже есть и для биосимиляров рекомбинантных человеческих инсулинов, о чем будет рассказано ниже.
Что касается биосимиляров аналогов инсулина, то на сегодняшний день только один подобный препарат одобрен Европейским агентством по лекарственным средствам (EMA) для использования в Европейском Союзе (под торговым названием Абасаглар®) и Агентством по контролю за пищевыми продуктами и лекарственными средствами (FDA) в США (под торговым названием Басаглар®). При этом в США Басаглар был одобрен по упрощенной схеме (согласно ст. 505(b)(2) Федерального закона о пищевых продуктах, лекарственных и косметических средствах), то есть не как биосимиляр, а как «последующий» (follow-on) биологический препарат. Связано это с тем, что аналоги инсулина в настоящее время регистрируются в США как химические препараты, а не как биологические продукты. Регистрация аналогов инсулина как биопрепаратов начнется с 2020 года.
Программа разработки Басаглара включает два классических исследования III фазы в параллельных группах – ELEMENT 1 с участием больных сахарным диабетом (СД) 1 типа и ELEMENT с пациентами с СД 2 типа, однако до сих пор не проведено перекрестных исследований для сравнения с оригинальным инсулином гларгин. И хотя был представлен подгрупповой анализ результатов лечения пациентов, ранее получавших оригинальный инсулин гларгин, важно понимать, что он не был специально спланирован для оценки взаимозаменяемости препаратов. Таким образом, в настоящее время нет прямых доказательств в пользу взаимозаменяемости Басаглара и оригинального инсулина гларгин у пациентов с СД 1 и 2 типа.
Некоторые другие производители также разрабатывают биосимиляры аналогов инсулина, причем некоторые из них уже изучаются в исследованиях III фазы.
Нормативная база по биосимилярам
На международном уровне передовую роль в развитии нормативной базы по биосимилярам сыграло ЕМА, которое еще в начале 2005 года опубликовало свое первое всеобъемлющее руководство по данному вопросу. В рекомендациях EMA указано, что сходство биосимиляра с уже утвержденным референтным биологическим препаратом должно быть продемонстрировано с помощью оценки качества, выполнения доклинических и клинических исследований. Клинические фармакокинетические и фармакодинамические исследования рассматриваются ЕМА как достаточно чувствительные, чтобы показать эквивалентность или различия между препаратами. Дизайн соответствующих исследований зависит от таких факторов, как клинический контекст, безопасность и фармакокинетические характеристики референтного продукта, выбор клинически значимых фармакодинамических маркеров. Кроме того, особое внимание при регистрации биосимиляров уделяется иммуногенности, которая оценивается, как правило, с помощью определения частоты выявления соответствующих антител и их титров. Такая оценка должна быть предусмотрена в рамках клинических исследований безопасности/сопоставимости биосимиляров. Их продолжительность определяется индивидуально в каждом случае, но должна быть не менее 12 мес для препаратов, которые применяются длительно. Накопленный до настоящего времени опыт свидетельствует о том, что нежелательные явления, возникающие в результате иммуногенности некоторых биопрепаратов, в том числе биосимиляров, являются редкими и поэтому могут быть не обнаружены в относительно небольших и/или краткосрочных исследованиях. Также для оценки иммуногенности очень актуален непрерывный послемаркетинговый мониторинг безопасности.
Кроме того, EMA также разработало 12 отдельных руководств, описывающих особенности проведения клинических исследований для различных классов биопрепаратов, таких как моноклональные антитела (МАт), интерферон-α и -β, гранулоцитарный колониестимулирующий фактор, эритропоэтин, фолликулостимулирующий гормон, гормон роста, низкомолекулярные гепарины, человеческий инсулин и его аналоги.
В США развитие нормативной базы по биосимилярам происходило более медленно. Принятый в 2009 году Закон о ценовой конкуренции и инновациях биологических лекарств разрешает сокращенную процедуру регистрации биологических препаратов, являющихся подобными препаратам, ранее уже одобренным FDA. В феврале 2012 года FDA выпустило соответствующее руководство «Научные аспекты подтверждения биоаналогичности по отношению к референтному препарату сравнения», пересмотренное затем в 2015 году.
Всемирная организация здравоохранения (ВОЗ) в 2009 году опубликовала свое «Руководство по оценке аналогичных биотерапевтических продуктов», которое было основано на опыте и соответствующих руководствах ЕС.
Взаимозаменяемость биосимиляров
ЕМА не комментирует взаимозаменяемость биосимиляров и оставляет решение о взаимозаменяемости и/или автоматическом замещении «национальным компетентным органам». Позиция некоторых из них обсуждается ниже. Тем не менее следующее заявление ЕМА свидетельствует о его позиции против идеи автоматического замещения биосимилярами без участия лечащего врача: «Так как биосимиляры и референтные биологические лекарственные средства похожи, но не идентичны, выбор между ними должен осуществляться на основании заключения квалифицированного медицинского работника».
FDA в отличие от EMA определяет некоторые стандарты взаимозаменяемости и предусматривает возможность автоматического замещения на биосимиляры на уровне аптек. Многие американские штаты рассматривают или уже ввели соответствующие законы.
Международными регулирующими органами пока не представлено конкретных рекомендаций по проведению исследований для подтверждения взаимозаменяемости биосимиляров с оригинальными препаратами. В то же время уже есть прецеденты проведения некоторыми производителями биосимиляров перекрестных исследований, например, с рекомбинантным человеческим гормоном роста (Омнитроп и Валтропин) и эпоэтином (Ретакрит).
Так, у детей с идиопатическим дефицитом гормона роста Омнитроп показал эффективность и безопасность, сопоставимые с оригинальным рекомбинантным человеческим гормоном роста Генотропином. В перекрестном исследовании Romer и соавт. (2009) 89 детей с соматотропной недостаточностью, ранее не получавшие лечения по поводу этой патологии, были рандомизированы для применения препаратов Омнитроп или Генотропин в течение 9 месяцев. Затем пациенты группы Генотропина были переведены на Омнитроп, в результате чего все участники исследования в течение примерно 7 последующих лет получали Омнитроп. В обеих группах пациентов были получены одинаковые результаты, что позволило сделать вывод о клинической сопоставимости биосимиляра и оригинального препарата. Позже был проведен post hoc анализ, в котором сравнили результаты лечения пациентов, которые были переведены с Генотропина на биосимиляр, и пациентов, у которых лечение сразу было начато с Омнитропа (Romer et al., 2011). Этот анализ показал отсутствие различий между группами по эффективности, безопасности и иммуногенности. Похожие данные получены и в других работах с переводом пациентов на Омнитроп (Flodmark et al., 2013; Rashid et al., 2014).
В двойном слепом перекрестном исследовании III фазы, проведенном Wizemann и соавт. (2008), биосимиляр эпоэтин-зета показал терапевтическую эквивалентность референтному эпоэтину-альфа в поддержании целевых уровней гемоглобина. Пациенты получали эпоэтин-зета или эпоэтин-альфа внутривенно в течение 12 недель, а затем альтернативное лечение на протяжении еще 12 недель. Средние уровни гемоглобина у пациентов, получавших эпоэтин-зета и эпоэтин-альфа, находились в пределах заранее установленного диапазона эквивалентности, при этом не наблюдалось никаких неожиданных нежелательных явлений.
В исследовании Sokka и соавт. (2015) 39 пациентов были переведены с оригинального препарата инфликсимаба (моноклональные антитела к фактору некроза опухоли альфа) Ремикейд на биосимиляр Инфлектра. В среднем через 11 мес после перевода на биосимиляр не было отмечено существенных изменений в выраженности симптомов, активности заболевания по оценкам пациентов и врачей, а также частоте нежелательных явлений. Эффективность и безопасность перевода пациентов с оригинального инфликсимаба на другой биосимиляр Ремсима в настоящее время оценивается в еще одном исследовании NOR-SWITCH (прим. ред. – его результаты уже опубликованы и показали сопоставимость препаратов Ремсима и Ремикейд).
Взаимозаменяемость vs автоматического замещения
Если биосимиляр демонстрирует взаимозаменяемость с оригинальным препаратом, возникает вопрос о возможности и экономической целесообразности автоматического замещения. Во многих странах разрешено замещение низкомолекулярных оригинальных препаратов соответствующими генериками, причем оно возможно даже в случае препаратов с узким терапевтическим диапазоном или высокой активностью (например, иммунодепрессантов). Согласно позиции ВОЗ решение о разрешении автоматического замещения оригинальных биопрепаратов биосимилярами следует принимать на национальном уровне, при этом их взаимозаменяемость должна быть подтверждена соответствующими научными и клиническими данными.
В некоторых европейских странах в настоящее время разрешается замещение биосимилярами при определенных обстоятельствах, в других этот вопрос только обсуждается.
Так, например, законодательство Франции позволяет замену фармацевтами оригинального биопрепарата на биосимиляр, но только при соблюдении ряда условий: пациент только начинает курс лечения (перевод с оригинального препарата не разрешен); биосимиляр принадлежит к той же группе, что и референтный препарат; лечащий врач явно не исключил возможность такой замены.
В Норвегии с 2015 года признана взаимозаменяемость биосимиляра Ремсима с оригинальным инфликсимабом, а региональные органы здравоохранения стараются перевести на препарат Ремсима как можно больше пациентов для сокращения затрат (биосимиляр на 69% дешевле референтного препарата).
Позиция Комиссии по оценке лекарственных средств Нидерландов заключается в том, что неконтролируемой замены биологических лекарственных средств (независимо от того, являются ли они инновационными продуктами или биосимилярами) следует избегать, и что решение о переводе пациента с одного биопрепарата на другой должно приниматься с участием лечащего врача и клинического фармацевта.
Финское агентство по лекарственным средствам допускает взаимозаменяемость биосимиляров и референтных препаратов под контролем специалиста здравоохранения.
В Германии в том случае, когда в рецепте на биопрепарат указано международное непатентованное название (МНН), фармацевту рекомендуется проконсультироваться с лечащим врачом относительно выбора.
Как уже было отмечено, в США допускается возможность автоматического замещения (без участия лечащего врача) оригинальных биопрепаратов биосимилярами с доказанной взаимозаменяемостью. Однако на момент написания статьи (январь 2016 года) только один продукт в США был лицензирован как биосимиляр – филграстим Зарсио, причем с пока еще недоказанной взаимозаменяемостью.
Дизайн исследований для оценки взаимозаменяемости аналогов инсулина
Для подтверждения биоподобности биосимиляра оригинальному аналогу инсулина ЕМА требует проведения ряда клинических исследований. Первым и важным шагом являются исследования I фазы, демонстрирующие аналогичные референтному продукту фармакокинетические и фармакодинамические профили. В то же время нет особой потребности в исследованиях эффективности, поскольку конечные точки, используемые в таких испытаниях (как правило, уровень HbA1c), не считаются достаточно чувствительными для обнаружения потенциально клинически значимых различий между двумя препаратами инсулина. Что касается исследований безопасности (III фазы), то в рекомендациях EMA подчеркивается, что они должны быть выполнены с особым акцентом на иммуногенность в качестве первичной конечной точки и включать достаточное количество больных СД 1 типа. Немаловажным является и постмаркетинговый фармаконадзор для исключения редких нежелательных явлений и подтверждения профиля безопасности (иммуногенности) биосимиляра.
Однако по мнению авторов, даже если было доказано подобие биосимиляра и оригинального аналога инсулина (а это означает, что продукт является безопасным и эффективным), таких данных недостаточно для подтверждения их взаимозаменяемости без проведения специально спланированных перекрестных исследований.
На момент написания статьи было опубликовано только одно перекрестное исследование по сравнению биосимиляра рекомбинантного человеческого инсулина с уже зарегистрированными препаратами (Segal et al., 2013). В этом многоцентровом открытом нерандомизированном интервенционном обсервационном исследовании пациентов с СД 1 и 2 переводили с одного из зарегистрированных предварительно смешанных человеческих инсулинов (Актрафан, Хумулин, Инсуман) на Биосулин (зарегистрирован как биосимиляр в ЮАР). Последний продемонстрировал эквивалентный гликемический контроль без каких-либо новых или серьезных побочных эффектов.
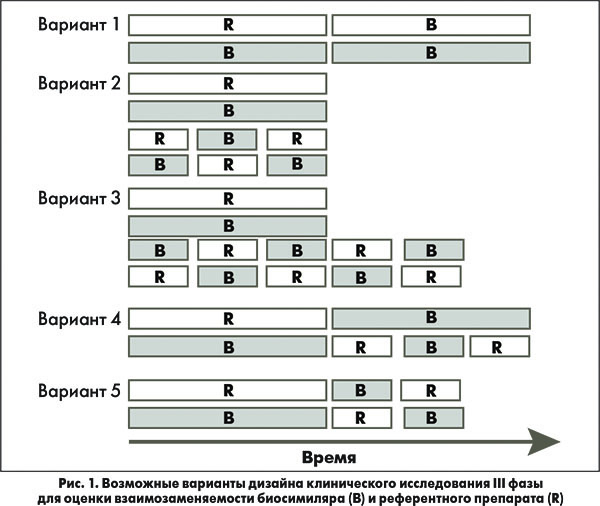
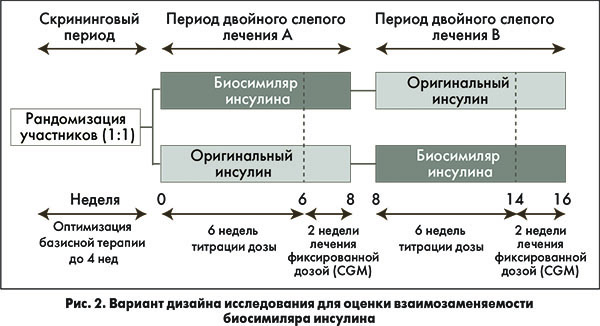
Несколько вариантов дизайна исследований по оценке взаимозаменяемости биосимиляров показаны на рисунке 1, а на рисунке 2 представлена гипотетическая схема исследования для демонстрации взаимозаменяемости биосимиляра инсулина.
В дополнение к систематическим клиническим перекрестным исследованиям целесообразно также проведение долгосрочных испытаний в условиях реальной клинической практики в более крупных и более разнообразных популяциях пациентов для оценки долгосрочной безопасности и клинической эффективности биосимиляров.
Взаимозаменяемость в клинической практике
Внедрение принципа взаимозаменяемости биосимиляров в клиническую практику может быть связано с неожиданными нежелательными явлениями. Многочисленные факторы (возраст, пол, длительность заболевания, наличие хронических осложнений и сопутствующих заболеваний) могут сыграть определенную роль при замещении одного препарата инсулина другим. С этой точки зрения представляется целесообразным не рекомендовать автоматическое замещение на уровне аптек, пока соответствующие клинические испытания не покажут отсутствие существенного влияния подобного замещения на эффективность или безопасность терапии.
Поскольку биосимиляры аналогов инсулина будут доступны в ближайшее время и существует неопределенность в отношении их использования, диабетологические организации, в частности Diabetes UK, разрабатывают свои рекомендации по их применению. Так, согласно рекомендациям Diabetes UK инсулинотерапию следует начинать с препаратов человеческого инсулина, при необходимости переводя пациентов на аналоги инсулина для достижения оптимального контроля заболевания. Биосимиляры инсулинов можно использовать только с согласия пациента после четкого разъяснения ему преимуществ и рисков. Кроме того, Diabetes UK рекомендует не менять оригинальный аналог инсулина на биосимиляр без серьезной клинической причины у пациентов с хорошо контролируемым уровнем глюкозы в крови. Diabetes UK также выступает за выписывание рецептов на аналоги инсулина под их торговым наименованием, а не МНН, чтобы избежать путаницы. Если перевод на биосимиляр необходим (например, в отделении неотложной помощи, для стационарных больных, во время путешествия и т.д.), рекомендуется снижение дозы инсулина примерно на 10% (с последующей коррекцией) и более частый контроль глюкозы капиллярной крови, что позволит сохранить гликемический контроль без повышенного риска развития гипогликемии. Следует рекомендовать пациентам сообщать о любых нежелательных явлениях, связанных с применением нового препарата.
В США недавно был проведен опрос пациентов об их гипотетической готовности перейти на менее дорогостоящий биосимиляр аналога инсулина (Wilkins et al., 2014). Он показал, что 66% пациентов «определенно» или «вероятно» готовы использовать биосимиляры аналогов инсулина, в то время как 17% сообщили, что «маловероятно» или «определенно не будут» их применять. Пациенты с СД 2 типа показали больше готовности использовать биосимиляры, чем пациенты с СД 1 типа.
Обеспокоенность пациентов была связана со сравнительной эффективностью бисимиляров аналогов инсулина и референтных препаратов, возможными побочными эффектами, необходимостью смены доставочного устройства (значительная доля респондентов заявила, что они будут рассматривать биосимиляр только в том случае, если он будет абсолютно идентичным их нынешнему препарату). Кроме того, многие пациенты заявили, что рассматривают определенный бренд в качестве гарантии качества, эффективности и надежности препарата.
Таким образом, в скором времени на фармацевтическом рынке будут доступны биосимиляры аналогов инсулина, что расширит возможности лечения пациентов с СД. Однако для рассмотрения вопроса о взаимозаменяемости с оригинальными препаратами необходимы дополнительные, в том числе перекрестные, исследования.
Сокращенный перевод с англ. Натальи Мищенко
Список литературы находится в редакции.
Diabetes, Obesity and Metabolism 18:
737-746, 2016.