


13 листопада, 2017
Патогенез и лабораторная диагностика синдрома диссеминированного внутрисосудистого свертывания в акушерстве
 В настоящее время акушерские кровотечения остаются основной причиной материнской смертности в мире [1]. Состоянием, которое приводит к кровотечениям с высокой смертностью при беременности, является диссеминированное внутрисосудистое свертывание (ДВС) крови.
В настоящее время акушерские кровотечения остаются основной причиной материнской смертности в мире [1]. Состоянием, которое приводит к кровотечениям с высокой смертностью при беременности, является диссеминированное внутрисосудистое свертывание (ДВС) крови.
Впервые ДВС описал в 1901 г. Joseph DeLee, который, наблюдая случай смертельного кровотечения при преждевременной отслойке нормально расположенной плаценты, объяснил это состояние как «временная гемофилия» [2]. Термин «ДВС-синдром» непосредственно предложил в 1950 г. американский патолог D. McKay. При вскрытии трупа женщины, погибшей на фоне отслойки плаценты от множественных массивных кровоизлияний, он обнаружил многочисленные тромбы, обтурирующие, главным образом, мелкие и мельчайшие сосуды. Позже, в 1965 г. D. McKay опубликовал монографию «Синдром диссеминированного внутрисосудистого свертывания как промежуточный механизм патогенеза болезней человека» [3]. Необходимо отметить, что D. McKay принадлежит одно из удачных определений ДВС-синдрома: «Это динамический биологический процесс, который вовлекает множество химических веществ и физиологических активаторов. Возникая в момент проникновения в кровь прокоагулянтного материала, он прогрессирует до стадии агрегации тромбоцитов и формирования фибрина, которые могут приводить к образованию микротромбов в капиллярах, артериолах и венах различных органов. Внутрисосудистое свертывание часто сочетается с активацией фибринолитической системы, расщеплением фибрина и фибриногена, высвобождением продуктов их деградации. Этот процесс сопровождается сильной вазомоторной реакцией и не заканчивается до тех пор, пока коагуляционный механизм и вазомоторный аппарат не нормализуют свою функцию, а продукты деградации фибрина/фибриногена не будут удалены из крови».
Современные представления о ДВС-синдроме позволяют заключить, что это – патологический синдром, в основе которого лежит активация сосудисто-тромбоцитарного и коагуляционного гемостаза. В результате этого кровь сначала сворачивается в микроциркуляторном русле, блокирует его фибрином и клеточными агрегатами, а при истощении потенциала свертывающей и противосвертывающей систем теряет способность к свертыванию, что проявляется кровотечением и развитием синдрома полиорганной недостаточности. ДВС-синдром, соответственно, является ключевым фактором, обусловливающим развитие полиорганной недостаточности и летальный исход по причине смертельного кровотечения. ДВС-синдром неспецифичен и универсален, так как возникает при самых разнообразных заболеваниях.
Патогенез ДВС-синдрома
Основа патогенеза ДВС-синдрома заключается в глубокой дисфункции всех звеньев системы гемостаза, которая характеризуется последовательной их активацией и истощением. По этой причине происходит смена фаз ДВС-синдрома от гиперкоагуляции и высокой спонтанной агрегации тромбоцитов в начале до переходного периода и последующей глубокой гипокоагуляции, вплоть до полной несвертываемости крови и тромбоцитопении в конце. Уже на ранних этапах наблюдается истощение не столько факторов свертывания, сколько важнейших физиологических антикоагулянтов – протеина С (ПрС), протеина S (ПрS), антитромбина III (АТIII). Угнетение физиологических антикоагулянтов тем значительнее, чем тяжелее ДВС. У больных с различными по происхождению вариантами ДВС-синдрома в процессе их формирования и развития отмечается ряд принципиальных закономерностей:
- наступление фазы гиперкоагуляции;
- срыв и прогрессирующее истощение основных антикоагулянтов (ПрС, ПрS, АТIII).
Стадии ДВС-синдрома
I стадия – гиперкоагуляция. При однократном и массивном образовании тромбопластина она кратковременна, но имеет отчетливые лабораторные признаки. Данный период характеризуется активацией плазменных систем свертывания крови, внутрисосудистой агрегацией тромбоцитов и других форменных элементов крови, нарушением микроциркуляции в разных органах в результате блокады сосудистого русла массами фибрина и агрегатами клеток.
II стадия – гипокоагуляция – обусловлена потреблением значительной части имеющихся в организме фибриногена, факторов V, VIII, XIII и других прокоагулянтов, а также тромбоцитов. Одновременно в крови накапливаются патологические ингибиторы свертывания крови, в частности продукты деградации фибрина и фибриногена (ПДФ), вызывающие увеличение антикоагулянтной активности крови.
III стадия – активация фибринолиза – афибриногенемии с патологическим фибринолизом. Активация фибринолитической системы ведет к растворению кровяных сгустков и создает предпосылки для развития геморрагического синдрома.
IV стадия – восстановительная. Характеризуется возращением к физиологическим границам коагуляционного потенциала. В этой стадии в той или иной мере восстанавливаются функции органов в зависимости от степени их поражения. Стадия может закончиться полным выздоровлением. Возможно развитие тяжелых осложнений уже в отсутствие как такового ДВС-синдрома – почечная, печеночная недостаточность, неврологические, кардиальные и другие осложнения.
Причины ДВС-синдрома в акушерстве
ДВС-синдром зависит от характера акушерской патологии, вызвавшей кровотечение, сопутствующих соматических заболеваний, особенностей течения беременности и др. При этом наблюдается множество клинических и лабораторных вариантов ДВС-синдрома, протекающего индивидуально у каждой пациентки. Причинами ДВС-синдрома в акушерстве могут быть [4, 5]:
- отслойка плаценты;
- септический аборт и внутриутробная инфекция;
- эмболия околоплодными водами;
- внутриутробная гибель плода;
- внематочная беременность;
- преэклампсия/эклампсия;
- кесарево сечение (10%);
- трансфузия несовместимой крови;
- травматические роды, в том числе энергичный массаж матки (высвобождение тканевого фактора);
- синдром массивной гемотрансфузии;
- HELLP-синдром;
- острая жировая дистрофия печени.
Патофизиология ДВС-синдромa при акушерской патологии
В данном аспекте очень важным является понимание процесса свертывания крови в норме и наблюдаемого во время ДВС. Нормальный коагулянтный ответ начинается с воздействия тканевого фактора (TF) вместе с фактором VIIa на фактор Xа для дальнейшего превращения протромбина в тромбин (IIa) [6]. Тромбин является прокоагулянтом при превращении фибриногена в фибрин и контролирует антикоагуляционный процесс путем генерирования активированного ПрС (аПрС), снижая активность факторов Vа и VIIIа. Растворение сгустка и генерирование продуктов деградации фибрина/фибриногена происходят через тромбининдуцированный тканевой активатор плазминогена (t-PA) с регуляцией фибринолиза, включающие активацию ингибитора фибринолиза тромбином (TAFI) [7,8]. Таким образом, тромбин играет центральную роль в балансе между про- и антикоагулянтной функциями, а также про- и антифибринолитической активностями (рис. 1).
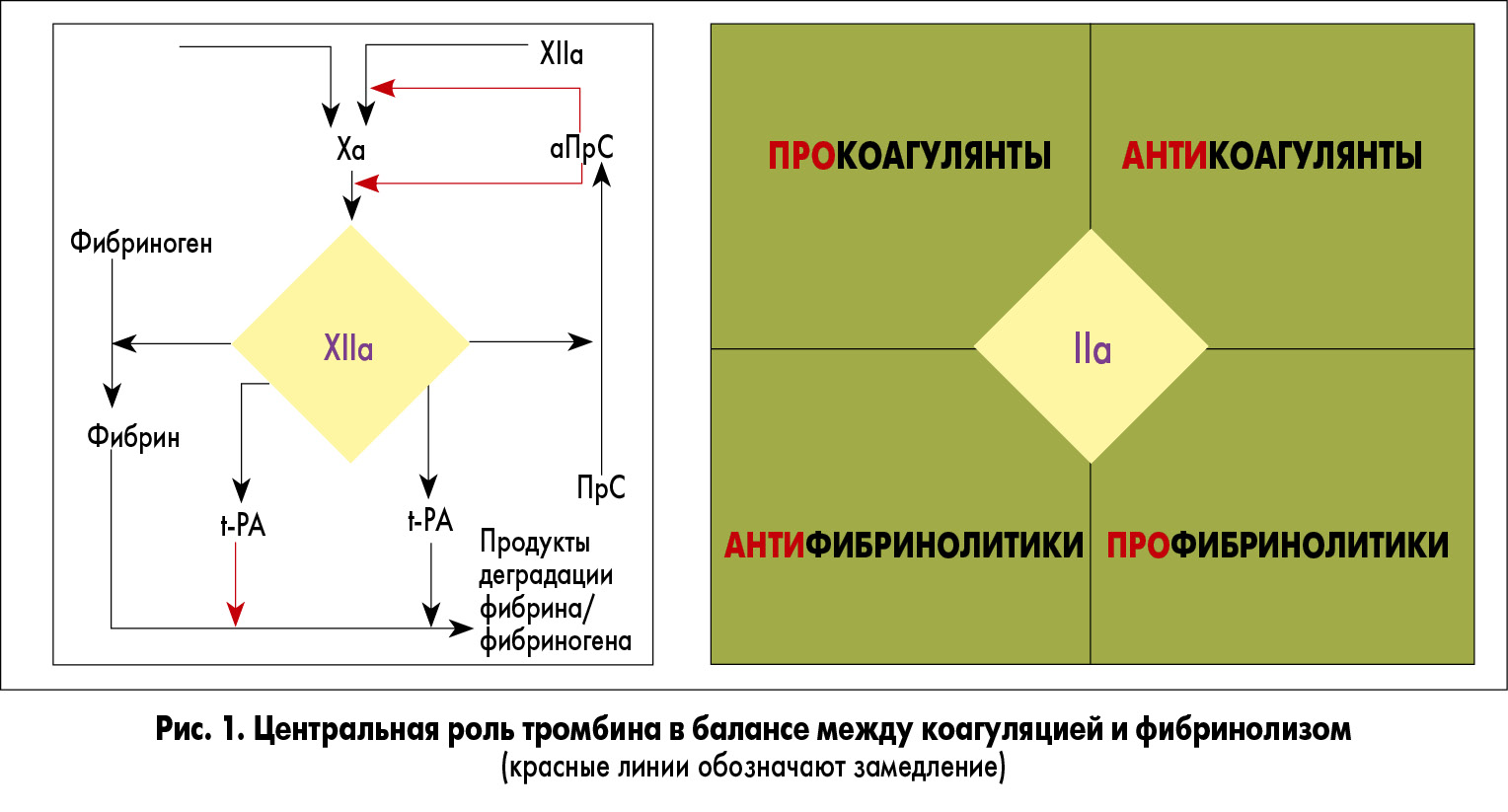
Важное место в развитии ДВС-синдрома принадлежит тромбину, для которого характерна чрезмерная генерация in vivo. Хотя образование тромбина в основном зависит от протромбиназного комплекса на поверхности тромбоцитов, однако клетки без фосфолипидов также могут поддерживать такие реакции in vivo [9, 10]. Они формируются в результате апоптоза или повреждения клеточных мембран, выпячивая фосфатидилсерин, находящийся на внутренней стороне мембраны. Микрочастицы, которые переносят внутренний фосфатидилсерин, продуцируют прокоагулянт, и их уровни повышаются в период беременности [11].
Важное значение имеет также обеспечение фосфолипидной поверхности липопротеинами, такими как окисленные липопротеины низкой плотности и липопротеины очень низкой плотности. Содержание последних может увеличиваться при ДВС-синдроме в несколько раз [12]. Кроме того, дисрегуляция липопротеинов влияет на активность тромбина через относительное снижение уровня липопротеинов высокой плотности, обладающих антикоагулянтными свойствами [13]. Показано, что циркулирующие липопротеины коррелируют с более высокой частотой преэклампсии [14]. У женщин с преэклампсией отмечены 3-кратное повышение уровня липопротеинов очень низкой плотности и значительно сниженная концентрация липопротеинов высокой плотности. Этот дисбаланс между про- и антикоагулянтными липопротеинами приводит к эндотелиальной дисфункции и служит патогенезом преэклампсии.
Плацента как активатор коагуляционной системы
Плацента обладает повышенным статусом коагуляционной активности за счет повышения продуцирования тканевого фактора. При этом наблюдается увеличение превращения протромбина (фактор II) в тромбин (фактор IIa) и дальнейшее расщепление фибриногена до фибрина. Повышается концентрация TAFIa, который вместе с повышенными уровнями ингибиторов активации плазминогена (PAI-1 и PAI-2) снижает фибринолитическую активность. Этот процесс обычно осуществляется через нормальную t-PA-индуцированную генерацию плазмина из плазминогена, в конечном итоге образуются ПДФ (рис. 2).
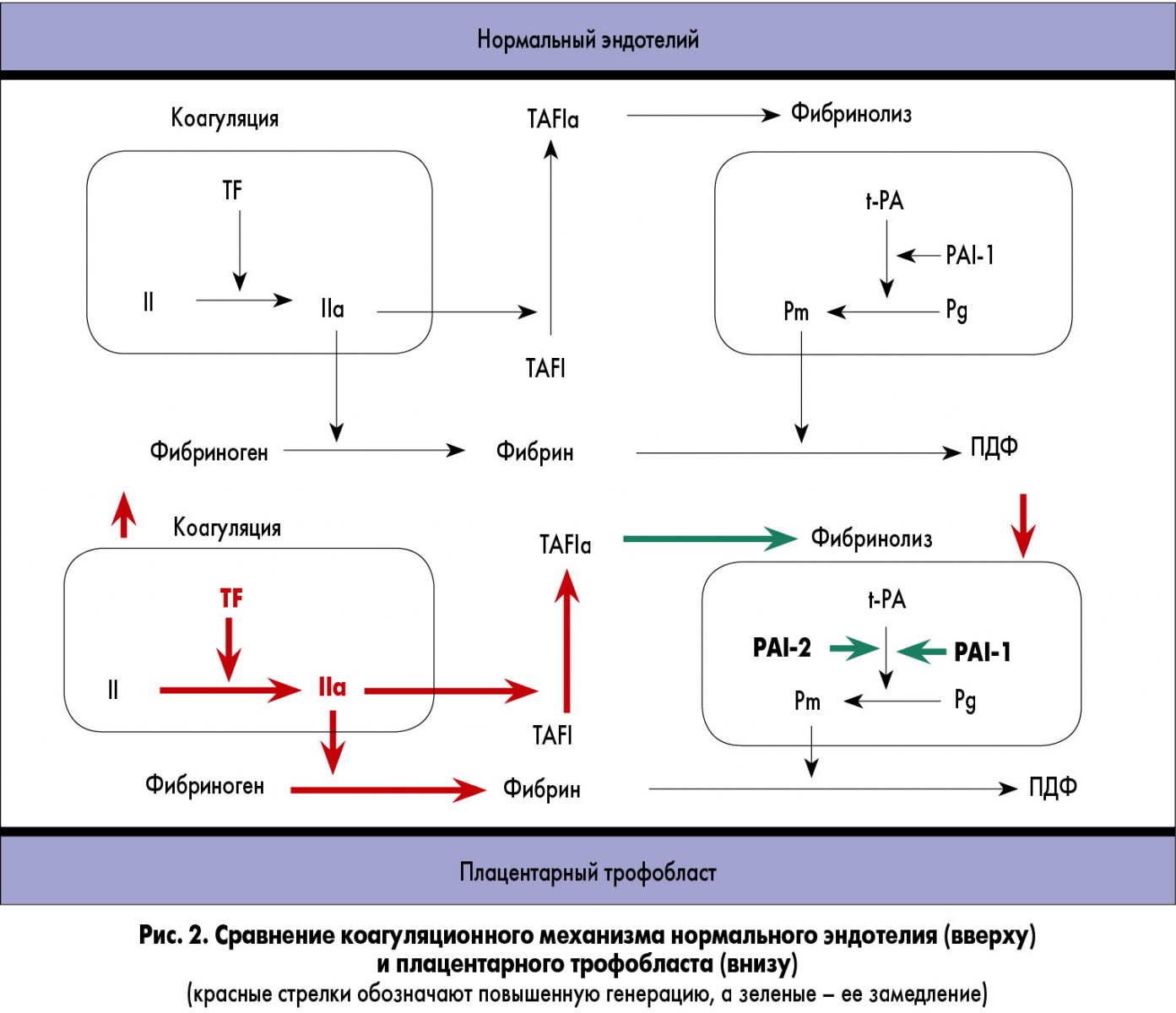
В сосудистой оболочке плаценты содержатся эмбриональные трофобластные клетки, обладающие клеточной способностью эндотелия регулировать гемостаз. Эти клетки обладают несколькими различными гемостатическими свойствами, которые важны для поддержания гемостаза при нормальной беременности. К ним относятся экспрессия TF, изменение антикоагулянтной функции, супрессия фибринолиза и воздействие анионных фосфолипидов.
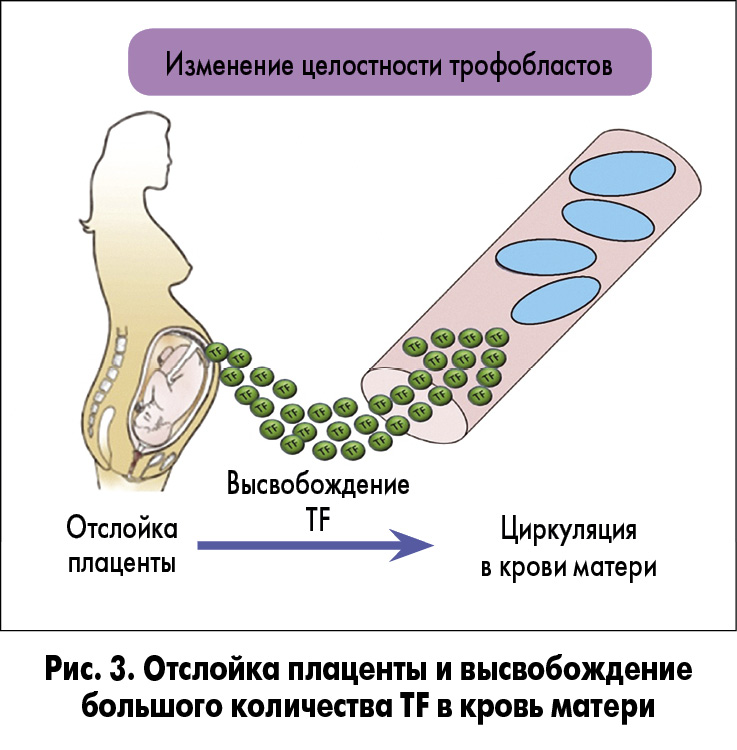
1. Экспрессия тканевого фактора. Синцитиотрофобласты нормальной плаценты человека обладают сильно выраженной активностью TF [15]. Нарушение целостности трофобластов – классическое явление при отслойке плаценты – приводит к высвобождению большого количества TF в материнское кровообращение. Это способствует активации коагуляционного каскада и распространению воспалительного ответа, который может легко стать системным, что приведет к неконтролируемому высвобождению тромбина и последующему развитию ДВС [16] (рис. 3). Для поддержания гемостаза во время эмбрионального развития необходим соответствующий баланс между TF и его ингибитором (TFPI) в разных органах [17].
2. Изменение антикоагулянтных функций. Тромбомодулин экспрессируется на трофобластах плаценты таким же образом, как и на поверхности кровеносных сосудов [18]. В работе M. Boffa и соавт. [19] показано, что уровни растворимого тромбомодулина на 12-й неделе гестации были одинаковыми при нормальной и патологической беременности. Эндотелиальный рецептор ПрС (EPCR) также экспрессируется на синцитиотрофобластах, что дает возможность аПрС-зависимому протеаза-активированному рецептору-1 блокировать апоптоз клеток плаценты [20]. Высокие уровни антител к EPCR связаны с более высоким риском смерти плода. Аутоантитела (анти-EPCR) могут активировать комплемент и быть причиной провоспалительного разрушения трофобластов и гибели плода. Активность ПрС не зависит от сроков гестации, в то время как прогрессивное снижение уровня ПрS наблюдается с увеличением срока гестации. Резистентность активированного ПрС увеличивается в течение беременности у 45% беременных по сравнению с небеременными того же возраста [21]. Уровни АТIII не изменяются на протяжении беременности [22]. В целом системная антикоагулянтная активность снижена по сравнению с таковой у небеременных, что подтверждается общим прокоагулянтным сдвигом при нормальной беременности [23]. Отмечено закономерное снижение уровня АТIII при ДВС-синдроме из-за его действия на нейтрализацию тромбина, фактора Xa и других плазменных сериновых протеаз.
3. Угнетение фибринолиза. При увеличении содержания PAI-1 плацента продуцирует PAI-2. В случае нормальной беременности уровень PAI-1 увеличивается постепенно, достигая заметного повышения в III триместре. Это значительное повышение происходит без относительных изменений уровня t-PA и приводит к уменьшению лизиса сгустка и протромботического смещения у беременных [24]. Эта так называемая «повышенная защита» против лизиса сгустка действует опосредованно через ингибитор фибринолиза, активируемый тромбином. TAFI представляет собой карбоксипептидазу В-подобного проферемента, который синтезируется в печени и активируется тромбин-тромбомодулиновым комплексом. При активации снижается регуляция фибринолиза и значительно увеличивается уровень TAFI у беременных, достигая пика в последнем триместре. При ДВС-синдроме чрезмерная генерация тромбина еще больше повышает уровень TAFI как ингибитора фибринолиза.
4. Воздействие анионных фосфолипидов. Показано, что фосфолипиды играют важную роль в росте поверхности плаценты при дифференциации и внутриклеточном слиянии ворсистого цитотрофобласта в синцитиотрофобласт [25]. Экстернализация фосфатидилсерина является важным компонентом этого процесса интертрофобластного слияния. Дифференциация цитотрофобластных ворсинок – это результат перераспределения мембранных фосфолипидов с обогащенным фосфатидилсерином на поверхности синцитиотрофобласта. При патологических состояниях в акушерстве (ДВС) наблюдается насыщение фосфатидилсерином поверхности трофобласта.
Эндотелиальная дисфункция и активация тромбоцитов
Интактные, дисфункциональные или активированные клетки, а также остатки клеточных мембран, медиаторы воспаления и коагуляционные белки являются частью сложного механизма взаимодействия, в котором неконтролируемая активация коагуляционного каскада приводит к ДВС. Эндотелиальные клетки, тромбоциты и в некоторых случаях лейкоциты могут участвовать в возникновении процесса, который приводит к ДВС. Это, как правило, происходит при высвобождении провоспалительных цитокинов, распространяющих активацию коагуляции или индуцирующих экспрессию TF на их мембране [26, 27]. Системный воспалительный ответ, который связан с увеличением содержания циркулирующих провоспалительных цитокинов, таких как фактор некроза опухоли (TNF) и интерлейкин-1 (IL-1), приводит к сверхэкспрессии TF лейкоцитами и эндотелиальными клетками. Результатом этого будет неконтролируемый коагуляционный ответ, который, в конечном итоге, приведет к ДВС. TF может экспрессироваться не только мононуклеарными, но и сосудистыми эндотелиальными и опухолевыми клетками. Необходимо отметить, что если физиологические антикоагулянты функционируют нормально, то несмотря на мощное инициирование коагуляции TF, последняя может и не возникнуть. Однако при ДВС-синдроме происходит нарушение функции всех основных природных антикоагулянтов (АТIII, ПрС и TFPI). Концентрация АТIII, самого важного ингибитора тромбина, при ДВС заметно снижена, так как он расщепляется эластазой из активированных нейтрофилов. Действие антикоагулянтов связано с эндотелием, поэтому активация эндотелиальных клеток и их дисфункция являются важными компонентами дисбаланса между свертывающей и антисвертывающей системами крови.
Активация тромбоцитов может ускорить образование фибрина. Экспрессия TF в моноцитах заметно стимулируется в присутствии тромбоцитов и гранулоцитов в Р-селектинзависимых реакциях. Этот эффект является результатом индуцированной активации ядерного фактора каппа-В при связывании активированных тромбоцитов с нейтрофилами и мононуклеарными клетками. В период беременности лейкоциты матери имеют высокий активационный статус по сравнению с таковыми у небеременных и обладают характеристиками, как при сепсисе [28]. В случае сепсиса, вызванного инфекционными агентами, септическим абортом или эмболией амниотической жидкостью, это равновесие нарушено, и у матери развивается ДВС-синдром.
Лабораторная диагностика ДВС-синдрома
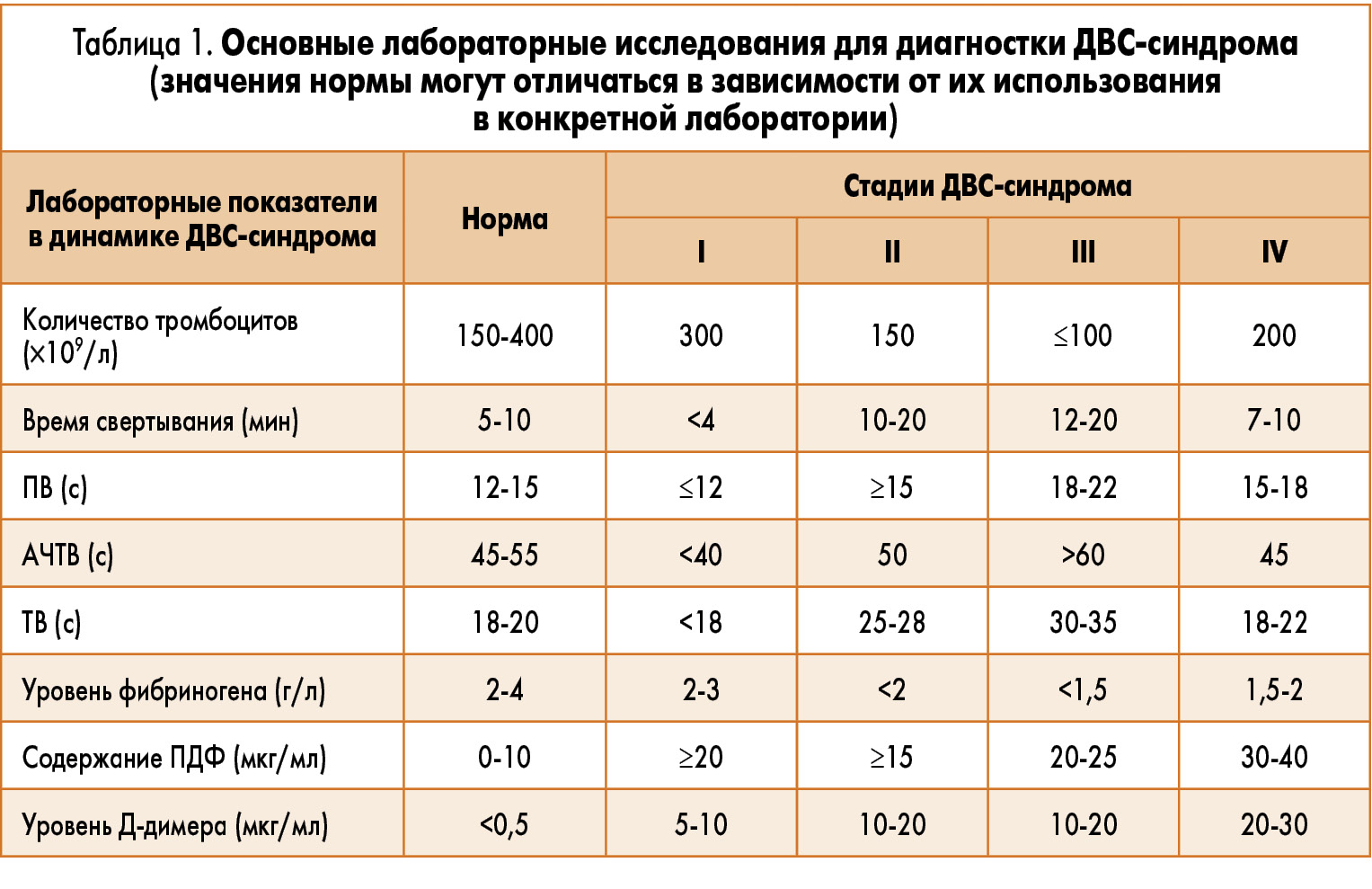
Лабораторная диагностика ДВС-синдрома должна быть срочной, основанной на четко разработанной системе простых и легко выполнимых тестов. Эти тесты также должны отражать фазы развития ДВС-синдрома и служить контролем проводимому лечению. Продолжительность клинических проявлений ДВС-синдрома может достигать 7-9 ч и более. Изменения в системе гемокоагуляции, определяемые с помощью лабораторных методов, сохраняются дольше, чем клинические, поэтому лабораторная диагностика ДВС-синдрома имеет первостепенное значение. Для лабораторной диагностики ДВС-синдрома используют простые и легкодоступные коагуляционные тесты: анализ количества тромбоцитов, время свертывания, протромбиновое время (ПВ), активированное частичное протромбиновое время (АЧТВ), тромбиновое время (ТВ), уровень фибриногена, ПДФ и Д-димера (табл. 1). Одним из ключевых моментов является то, что эти тесты отражают динамические изменения на основе клинических наблюдений.
Кроме основных лабораторных тестов врачу-клиницисту важно иметь информацию о возможности применения в своей практике более широкого спектра лабораторных исследований.
1. Дополнительные тесты. Уровни природных антикоагулянтов, таких как АТIII и ПрС, у 90% пациентов с ДВС-синдромом снижены. Лабораторные значения этих природных ингибиторов предоставляют ценную информацию для диагностики и мониторинга ДВС-синдрома, однако они не используются в лабораторных исследованиях, так как не добавляют никакой диагностической информации. Низкие уровни АТIII и ПрС не являются специфичными для ДВС-синдрома по причине того, что они могут быть связаны с заболеванием печени или с другими патологиями.
2. Новые методы исследования. Дополнительно к анализу общего количества тромбоцитов можно использовать другие параметры тромбоцитов для постановки диагноза ДВС-синдрома. Некоторые современные гематологические анализаторы имеют возможность идентифицировать «ретикулярные» тромбоциты, измеряемые как фракция незрелых тромбоцитов. Недавние исследования показали, что увеличение содержания «ретикулярных» тромбоцитов коррелирует с диагнозом явного ДВС-синдрома. Наличие «ретикулярных» тромбоцитов коррелирует с увеличением содержания ПДФ и является более точным прогностическим фактором ДВС, чем низкий уровень тромбоцитов. Поскольку ДВС включает активацию воспалительного процесса, анализ воспалительных маркеров, таких как С-реактивный белок, IL-6, IL-8, TNF, прокальцитонин и липопротеинсвязывающий белок, будут иметь широкое применение в самое ближайшее время.
3. Специализированные тесты. Для установления диагноза ДВС-синдрома могут применяться специализированные лабораторные исследования:
1) избыточная генерация тромбина:
- увеличение содержания комплекса тромбин-антитромбин;
- повышение уровня фибринопептидов;
- увеличение содержания фрагментов протромбина-1 и -2;
2) снижение уровней ПрС и ПрS, АТIII; повышенный фибринолиз:
- увеличение содержания плазмина;
- снижение уровня плазминогена;
- снижение концентрации α2-антиплазмина;
- увеличение содержания комплексов плазмин-антиплазмин;
- высокие уровни ингибиторов активатора плазминогена;
3) новые маркеры (тромбоз-воспаление);
- повышенный уровень растворимого тромбомодулина;
- увеличение количества гистонов и внеклеточной ДНК;
- увеличение количества групп белков высокой мобильности-1;
- активация нейтрофилов;
- снижение содержания ADAMTS-13 (a disintegrin and metalloprotei-nase with a thrombospondin type 1 motif, member 13);
- маркеры комплемента (С3, комплекс мембранной атаки и маннозосвязывающий лектин);
- пресепсин (растворимый кластер дифференцировки, субтип 14).
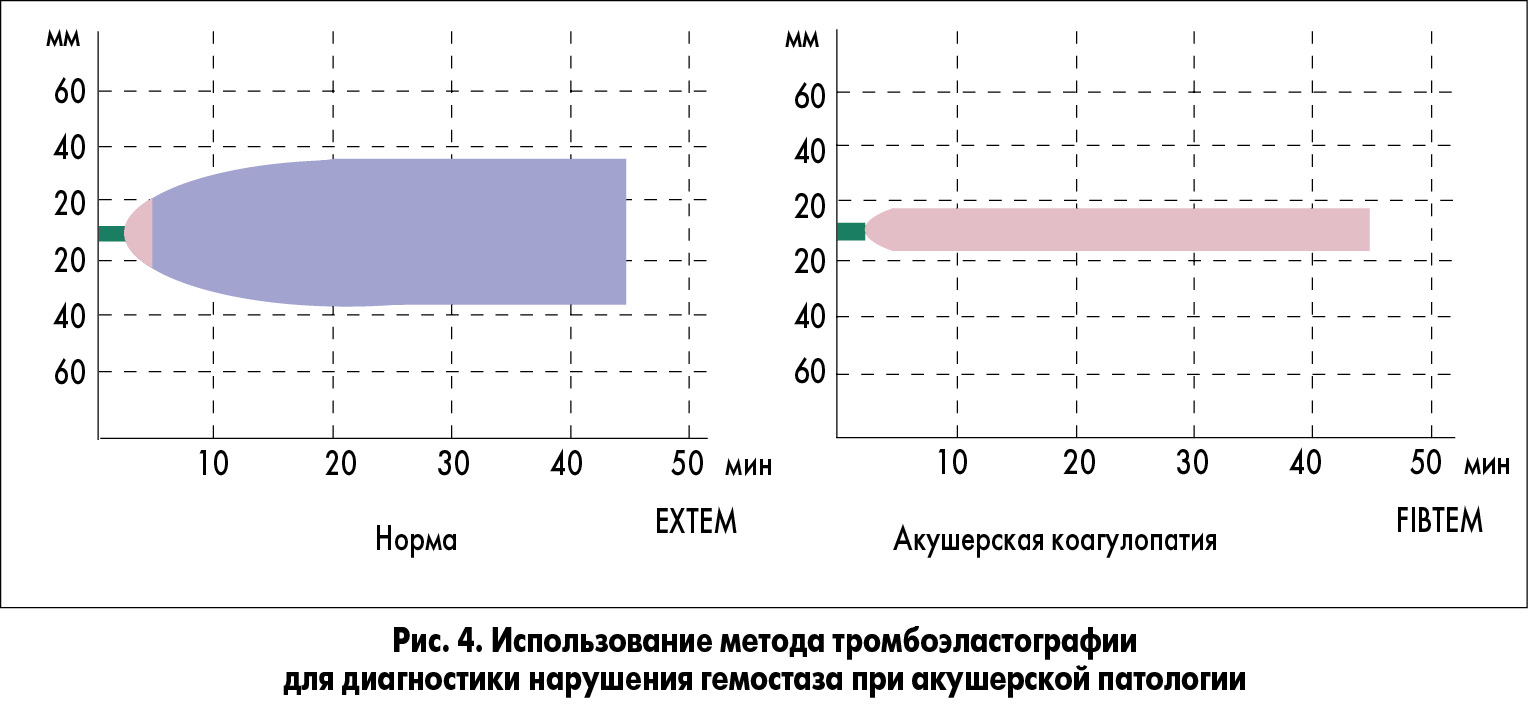
4. Тромбоэластография представляет собой интегральный метод оценки системы гемостаза с использованием анализа вязкоэластических свойств сгустка. Одновременная диагностика коагуляционного каскада, активности фибринолиза и тромбоцитарного звена может выявить расстройства системы гемостаза в течение 10-20 мин (рис. 4). Для этих целей широкое применение получили аналиазторы TEG (Haemoscope Corporation, США) и ROTEM (Tem GmbH, Германия).
Шкалы для диагностки ДВС-синдрома
Раннее и точное распознавание ДВС-синдрома является решающим для эффективного лечения этого осложнения. К сожалению, в большинстве случаев диагноз ДВС-синдром основан на клинической оценке состояния пациента. Следует отметить, что не существует ни одного лабораторного или клинического теста, который был бы чувствительным и специфичным для диагностики ДВС. По этим причинам, а также из-за необходимости предоставить врачу-клиницисту данные по раннему выявлению ДВС, были предприняты попытки создания скриниг-систем, которые базируются на идентификации пациентов с высоким риском этого опасного осложнения.
У пациентов с подозрением на ДВС-синдром для постановки диагноза предлагается использовать следующие шкалы: International Society on Thrombosis and Hemostasis (ISTH), Japanese Ministry of Health and Welfare (JMHW), Japanese Association for Acute Medicine (JAAM; табл. 2) [29-32].
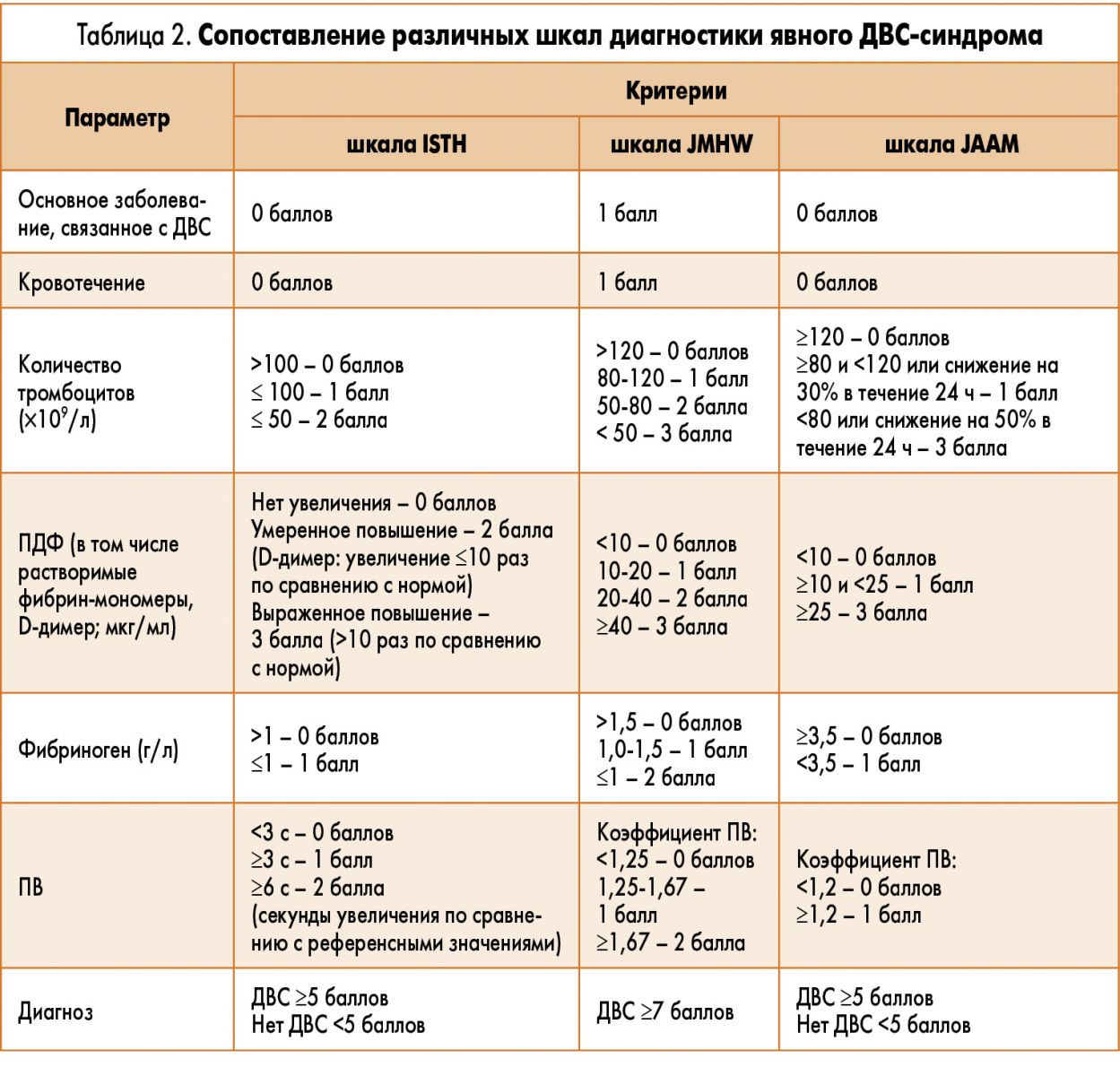
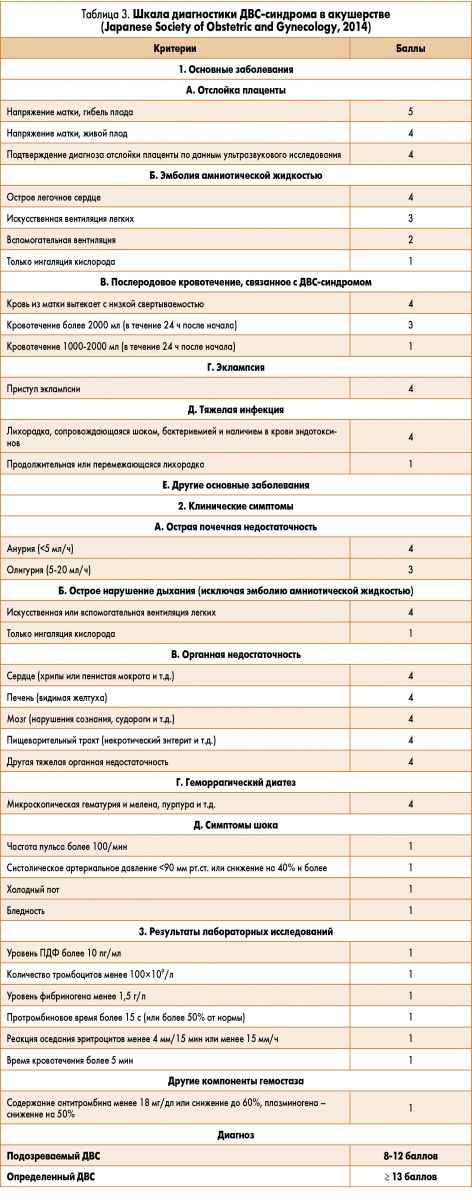
Общепринятые шкалы диагностики ДВС-синдрома по ISTH, JMHW, JAAM не всегда могут учитывать особенности физиологии гемостаза при беременности (увеличение содержания ПДФ, фибриногена) и особенности критических состояний в акушерстве. Для диагностики ДВС-синдрома в акушерстве целесообразно использовать шкалу Japanese Society of Obstetric and Gynecology (2014; [33]; табл. 3), которая учитывает особенности основного заболевания, клинических симптомов, органной недостаточности и лабораторных исследований. Эта шакала охватывает клинические ситуации с преобладанием кровотечения, микротромбоза и органной недостаточности. Несмотря на некоторую ограниченность, характерную для всех шкал, именно подобный комплексный подход должен использоваться врачом акушером-гинекологом и врачом анестезиологом-реаниматологом для диагностики ДВС-синдрома и выбора лечебной тактики: заместительной терапии компонентами крови или физиологическими антикоагулянтами.
Заключение
ДВС-синдром связан с акушерской патологией и представляет собой опасное для жизни состояние. Понимание механизмов развития и быстрая, информативная диагностика этой патологии, а также своевременное лечение способствуют благоприятному исходу. Лабораторные тесты являются очень важными компонентами диагностического процесса, однако ни один из них, доступный в настоящее время, не позволяет со 100% точностью подтвердить диагноз ДВС. Для диагностики ДВС-синдрома в акушерстве рекомендуется использовать пять основных исследований: определение количества тромбоцитов, уровней фибриногена, ПДФ, Д-димера, ПВ, а также современные тесты – выявление маркеров воспаления и метод тромбоэластографии. Для повышения диагностической значимости этих исследований при установлении диагноза ДВС-синдрома, связанного с акушерской патологией, применяют международные шкалы.
Литература
- Williams J., Mozurkewich E., Chilimigras J., Van De Ven C. Critical care in obstetrics: pregnancy-specific conditions. Best Pract Res Clin Obstet Gynaecol 2008; 22(5): 825-46.
- DeLee J.B. A case of fatal hemorrhagic diathesis, with premature detachment of the placenta. Am J Obstet Dis Women Child 1901; 44: 785-92.
- McKay D.G. Disseminated Intravascular Coagulation. An Intermediary Mechanism of Disease. New York: Harper & Row, 1965.
- Rattray D.D., O’Connell C.M., Baskett T.F. Acute disseminated intravascular coagulation in obstetrics: a tertiary centre population review (1980 to 2009). J Obstet Gynaecol Can 2012; 34: 341-347.
- Erez O., Novack L., Beer-Weisel R. et al. DIC score in pregnant women – a population based modification of the International Society on Thrombosis and Hemostasis score. PLoS One 2014; 9: e93240.
- Hoffman M., Monroe D.M. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am 2007; 21: 1-11.
- Medved L., Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost 2003; 89: 409-19.
- Bajzar L., Morser J., Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 1996; 271: 16603-8.
- Rosing J., van Rijn J.L., Bevers E.M. et al. The role of activated human platelets in prothrombin and factor X activation. Blood 1985; 65: 319-32.
- Moyer M.P., Tracy R.P., Tracy P.B. et al. Plasma lipoproteins support prothrombinase and other procoagulant enzymatic complexes. Arterioscler Thromb Vasc Biol 1998; 18: 458-65.
- Bretelle F., Sabatier F., Desprez D. et al. Circulating microparticles: a marker of procoagulant state in normal pregnancy and pregnancy complicated by preeclampsia or intrauterine growth restriction. Thromb Haemost 2003; 89: 486-92.
- Dennis M.W., Downey C., Brufatto N. et al. Prothrombinase enhancement through quantitative and qualitative changes affecting very low density lipoprotein in complex with C-reactive protein. Thromb Haemost 2004; 9: 522-30.
- Griffin J.H., Kojima K., Banka C.L. et al. High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest 1999; 103: 219-27.
- Sattar N., Bendomir A., Berry C. et al. Lipoprotein subfraction concentrations in preeclampsia: pathogenic parallels to atherosclerosis. Obstet Gynecol 1997; 89: 403-8.
- Aharon A., Brenner B., Katz T. et al. Tissue factor and tissue factor pathway inhibitor levels in trophoblast cells: implications for placental hemostasis. Thromb Haemost 2004; 92: 776-86.
- Oyelese Y., Ananth C.V. Placental abruption. Obstet Gynecol 2006; 108: 1005-16.
- Pedersen B., Holscher T., Sato Y. et al. A balance between tissue factor and tissue factor pathway inhibitor is required for embryonic development and hemostasis in adult mice. Blood 2005; 105: 2777-82.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004; 114: 409-14.
- Boffa M.C., Valsecchi L., Fausto A. et al. Predictive value of plasma thrombomodulin in preeclampsia and gestational hypertension. Thromb Haemost 1998; 79: 1092-5.
- Isermann B., Sood R., Pawlinski R. et al. The thrombomodulin–protein C system is essential for the maintenance of pregnancy. Nat Med 2003; 9: 331-7.
- Clark P., Brennand J., Conkie J.A. et al. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79: 1166-70.
- Bremme K., Ostlund E., Almqvist I. et al. Enhanced thrombin generation and fibrinolytic activity in normal pregnancy and the puerperium. Obstet Gynecol 1992; 80: 132-7.
- Cerneca F., Ricci G., Simeone R. et al. Coagulation and fibrinolysis changes in normal regnancy. Eur J Obstet Gynecol Reprod Biol 1997; 73: 31-6.
- Robb A.O., Mills N.L., Din J.N. et al. Acute endothelial tissue plasminogen activator release in pregnancy. J Thromb Haemost 2009; 7: 138-42.
- Das M., Xu B., Lin L. et al. Phosphatidylserine efflux and intercellular fusion in a BeWo model of human villous cytotrophoblast. Placenta 2004; 25: 396-407.
- Levi M. Pathogenesis and management of peripartum coagulopathic calamities (disseminated intravascular coagulation and amniotic fluid embolism). Thromb Res 2013; 131 (Suppl 1): S32-4.
- Levi M., van der Poll T. Inflammation and coagulation. Crit Care Med 2010; 38: S26-34.
- Sacks G.P., Studena K., Sargent K., Redman C.W. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol 1998; 179: 80-6.
- Bakhtiari K., Meijers J.C.M., de Jonge E., Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med 2004; 32: 2416-21.
- Yanada M., Matsushita T., Suzuki M., Kiyoi H., Yamamoto K., Kinoshita T. et al. Disseminated intravascular coagulation in acute leukemia: clinical and laboratory features at presentation. Eur J Haematol 2006; 77: 282-7.
- Gando S., Iba T., Eguchi Y., Ohtomo Y., Okamoto K., Koseki K. et al. A multicenter, prospective validation of disseminated intravascular coagulation diagnostic criteria for critically ill patients: Comparing current criteria. Crit Care Med 2006; 34: 625-31.
- Sawamura A., Hayakawa M., Gando S., Kubota N., Sugano M., Wada T. et al. Application of the Japanese Association for Acute Medicine disseminated intravascular coagulation diagnostic criteria for patients at an early phase of trauma. Thromb Res 2009; 124: 706-10.
- Minakami H., Maeda, Fujii T. et al. Guidelines for obstertical practice in Japan: Japan Society of Obstertics and Gynecology (JSOG) and Japan Association of Obstetricians and Gynecologists (JAOG) 2014 edition. J Obstet Gynaecol Res 2014 Jun; 40(6): 1469-99.