


21 червня, 2018
ІІ Школа з нервово-м’язових захворювань і захворювань периферичної нервової системи
25-26 травня в м. Львові Українська асоціація нервово-м’язових захворювань і захворювань периферичної нервової системи вдруге на високому рівні провела освітній захід із міжнародною участю, присвячений проблемам діагностики та лікування нервово-м’язових хвороб і невропатій. Наукова програма була присвячена розгляду рідкісних патологій, які потребують більш пильної уваги фахівців.

Вступну лекцію під назвою «Нервово-м’язові захворювання (НМЗ): де ми є та куди йдемо» прочитав іноземний гість – професор Андоні Уртізбереа (Інститут міології, м. Париж, Франція). Інститут міології функціонує на базі відомого госпіталю Ла Сальпетрієр. У цьому закладі працював іще Гійом Дюшен (1806-1875) – французький невролог, який першим описав прогресуючу міодистрофію й на честь якого вона була названа. Сьогодні Інститут міології – найбільший у Європі осередок клінічної та науково-педагогічної роботи з проблеми НМЗ. На базі інституту проводиться всесвітньо відома літня школа міології (The Summer School of Myology), яку відвідали 600 фахівців за останні 20 років. Щороку близько 4000 стаціонарних і амбулаторних пацієнтів отримують тут високоспеціалізовану допомогу. Інститут є базою для проведення клінічних досліджень.
Професор А. Уртізбереа розпочав лекцію з історичного зіставлення. Ще 50 років тому лікарі не мали знань і досвіду, щоб діагностувати НМЗ, а пацієнти не мали надії. Революція розпочалася 1986 року, коли було відкрито перший ген, відповідальний за НМЗ, – ген дистрофіну, дефект якого зумовлює м’язову дистрофію Дюшена. Наразі відомо вже понад 600 генних локусів, пов’язаних із НМЗ. Лікарі мають на порядок більше інформації, але в цьому різноманітті дедалі важче орієнтуватися.
Випробовуються нові напрями терапії – фармакологічна, генна, клітинна. Проте на сьогодні лише для невеликої частини НМЗ існує патогенетичне лікування; для решти пацієнтів ми маємо надію на його відкриття.
Лектор докладно розглянув сучасну класифікацію НМЗ, яка ґрунтується на анатомічному принципі – за відділами нервово-м’язової одиниці, в яких відбувається порушення (рис.).
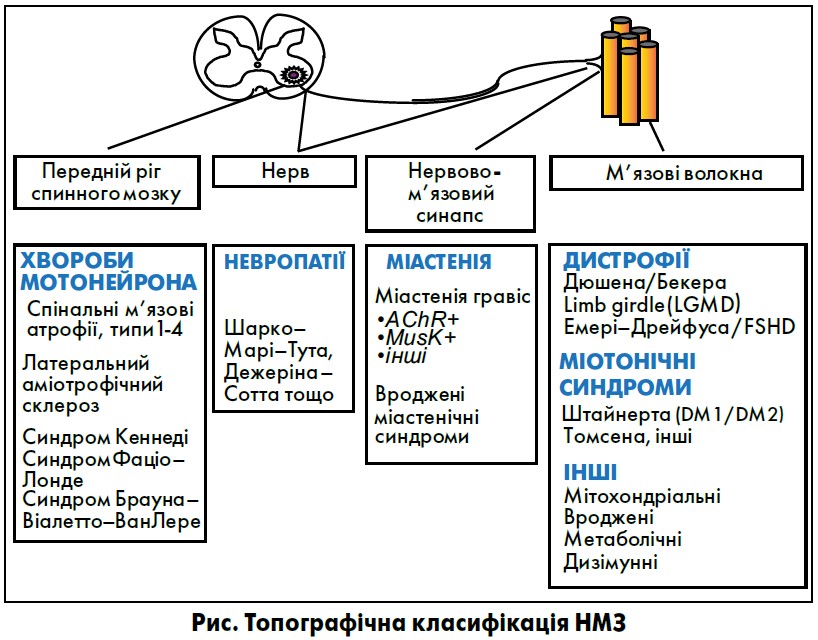 Найпоширеніші в популяції НМЗ, такі як міопатія Дюшена (МД), міотонічні дистрофії (тип 1 – хвороба Штайнерта), спінальні м’язові атрофії (СМА), є спадковими захворюваннями. Проте не всі НМЗ генетично детерміновані. Значна частка пацієнтів мають дизімунні, метаболічні, токсичні, інфекційні нейропатії та міопатії. І для них є більше можливостей лікування.
Найпоширеніші в популяції НМЗ, такі як міопатія Дюшена (МД), міотонічні дистрофії (тип 1 – хвороба Штайнерта), спінальні м’язові атрофії (СМА), є спадковими захворюваннями. Проте не всі НМЗ генетично детерміновані. Значна частка пацієнтів мають дизімунні, метаболічні, токсичні, інфекційні нейропатії та міопатії. І для них є більше можливостей лікування.
Залежно від того, де працює лікар і якої він спеціальності, він може мати вищу ймовірність зустріти те чи інше НМЗ, оскільки вони істотно відрізняються за віком маніфестації. Так, СМА, міотубулярна міопатія, МД 1 типу можуть мати антенатальні прояви; вроджені міопатії та м’язові дистрофії можна діагностувати при народженні, деякі НМЗ маніфестують у ранньому дитинстві (наприклад, СМА 1 та 2 типів), а деякі – в підлітковому чи дорослому віці (міопатія Беккера, хвороба Штайнерта, міастенія, кальпаїнопатія).
На запитання «Як часто можна зустріти НМЗ у своїй практиці?» немає чіткої відповіді, оскільки це залежить від конкретної популяції, що має специфічний генетичний склад. Лектор навів приклад Франції з населенням 65 млн мешканців, де діагностовано 4000 випадків дистрофінопатій (міопатії Дюшена, Беккера, в тому числі носії патологічних генів), 3500 випадків міотонічних дистрофій (типу 1 та 2), 3000 випадків FSHD (міопатії обличчя – лопатка – плече), 2500 випадків СМА всіх типів, 200 випадків кальпаїнопатії, 200 випадків хвороби
Помпе, 35 випадків осифікуючого міозиту й лише 1 випадок синдрому Брауна – Віалетто – Ван Лере.
Професор А. Уртізбереа закликав лікарів намагатися встановити точний діагноз, незважаючи на складність і високу вартість методів дослідження (насамперед генетичних), оскільки НМЗ відрізняються за прогнозом і підходами до лікування. Разом із тим, незважаючи на величезний прорив у молекулярно-генетичній діагностиці, обізнаний і досвідчений клініцист відіграє ключову роль.
Лектор озвучив десять «заповідей» фахівця з НМЗ (міолога):
– розпізнавайте хвороби, для яких існує лікування;
– зробіть усе можливе, щоб визначити мутацію молекулярно-генетичними методами;
– у разі позитивного результату забезпечте генетичне консультування;
– за потреби спрямуйте пацієнта до спеціалістів;
– будьте мультидисциплінарними, намагайтеся скласти цілісну картину;
– уникайте непотрібних досліджень;
– дотримуйтеся стандартів допомоги;
– пам’ятайте про профілактику;
– будьте чуйними;
– призначайте доказово обґрунтовану терапію.
Далі професор зупинився на семіотиці, нагадав кроки обстеження пацієнта з підозрою на НМЗ і найспецифічніші прояви патології. Під час збору анамнезу необхідно розпитати пацієнта чи батьків хворої дитини про скарги, вік та обставини початку захворювання, уточнити наявність таких симптомів, як слабкість, біль, судоми в м’язах (яких саме), порушення постави та ходи, швидкість прогресування симптомів. Також слід з’ясувати, чи приймає пацієнт препарати групи статинів.
 Сімейний анамнез – важлива складова обстеження, адже більшість НМЗ мають спадковий характер. Бажано скласти родовід пацієнта, звернувши увагу на можливість близькоспоріднених шлюбів у сім’ї, етнічні особливості. Важливо враховувати тип успадкування НМЗ: Х-зчеплене рецесивне (носіями є жінки, а хворіють лише чоловіки), аутосомно-домінантне (передаються від покоління в покоління), аутосомно-рецесивне (більш поширені в країнах зі спорідненими шлюбами).
Сімейний анамнез – важлива складова обстеження, адже більшість НМЗ мають спадковий характер. Бажано скласти родовід пацієнта, звернувши увагу на можливість близькоспоріднених шлюбів у сім’ї, етнічні особливості. Важливо враховувати тип успадкування НМЗ: Х-зчеплене рецесивне (носіями є жінки, а хворіють лише чоловіки), аутосомно-домінантне (передаються від покоління в покоління), аутосомно-рецесивне (більш поширені в країнах зі спорідненими шлюбами).
Під час огляду пацієнта необхідно оцінити поставу (гіперлордоз, сколіоз, інші деформації) та моторні функції: вставання (маневр Говера), піднімання рук (крилоподібні лопатки), ходу (симетрична чи ні, з підстрибуванням, розхитуванням), а також силу й тонус м’язів. У діагностиці деяких НМЗ і спадкової патології має значення обстеження обличчя (слабкість і асиметрія мімічних м’язів), язика, окорухової функції, стану шкіри.
Додаткові методи обстеження, найбільш важливі в нейроміології, – це електронейроміографія, візуалізація (комп’ютерна, магнітно-резонансна томографія), біопсія м’язів, біохімічні аналізи, ДНК-тестування. Проте лектор зазначив, що навіть у спеціалізованих центрах, таких як Інститут міології, біопсія відходить на задній план, поступаючись ДНК-діагностиці.
Генетичне тестування має першочергове значення для підтвердження діагнозу та призначення специфічної терапії. Поки що вона дорога й доступна не в усіх країнах, тому потребує співпраці лікарів із зарубіжними академічними центрами та лабораторіями. Крім того, генетична діагностика потребує участі фахівця-клініциста, який здатен критично оцінювати результати.
Щодо лікування НМЗ, то наразі більшість спадкових хвороб залишаються невиліковними. Допомога здебільшого зводиться до неспецифічної підтримувальної терапії та корекції (ортопедія, респіраторна підтримка). З метою профілактики нових випадків НМЗ у сім’ї проводять пренатальне тестування та генетичне консультування.
Методи генної та клітинної терапії поки що вивчаються в експериментальних дослідженнях. Але деякі НМЗ, наприклад хвороба Помпе, вже мають специфічне фармакологічне лікування*. З метою координації міжнародних зусиль у пошуку лікування НМЗ, нових малоінвазивних методів діагностики та створення реєстрів пацієнтів із рідкісними захворюваннями започатковано проект TREAT-NMD (Translational Research in Europe – Assessment and Treatment of Neuromuscular Diseases). До співпраці залучаються професійні асоціації, академічні та дослідні центри, фармацевтична індустрія, організації пацієнтів; передбачається також індивідуальне членство.
 Доктор медичних наук, професор Гаяне Рубенівна Акопян (ДУ «Інститут спадкової патології НАМН України», м. Львів) розповіла про молекулярні механізми лікування спадкових неврологічних хвороб і деякі перспективні терапевтичні засоби.
Доктор медичних наук, професор Гаяне Рубенівна Акопян (ДУ «Інститут спадкової патології НАМН України», м. Львів) розповіла про молекулярні механізми лікування спадкових неврологічних хвороб і деякі перспективні терапевтичні засоби.
При спадкових НМЗ прояви хвороби визначаються помилками в генетичній програмі – мутаціями. Хвороба виникає внаслідок абсолютного дефіциту білка чи неповноцінності його структури, в результаті чого білок не здатен виконувати свою функцію. Втрати кодуючих ділянок ДНК (делеції екзонів) типові для м’язових дистрофій та спінальних аміотрофій.
У здорових осіб виживання мотонейронів значною мірою залежить від білка SMN (Survival Motor Neuron), який синтезується з гена SMN1. Ген має 8 кодуючих ділянок ДНК – екзонів. У хворих на СМА делеція сьомого екзону гена SMN1 присутня в обох алелях гена (від матері й батька).
У результаті втрати генетичного матеріалу ген SMN1 неспроможний забезпечити синтез повноцінного білка SMN для підтримки функції виживання мотонейронів. У грудні 2016 року Управління з контролю якості продуктів харчування та лікарських засобів США (FDA) схвалило нусинерсен (Spinraza) – перший препарат для лікування дітей (у тому числі новонароджених) і дорослих зі СМА, викликаною мутацією в гені SMN1. Наразі для лікування СМА проводять клінічні випробування інших препаратів подібної дії (RG7916, Branaplan), а також цитопротектора нового покоління – Olesoxime, для яких є можливість перорального застосування.
Також вивчається можливість генної терапії – повного заміщення SMN1 шляхом трансфекції клітин за допомогою вірусних векторів (оболонок – носіїв генетичної інформації). Препарат Avexis перебуває на першому етапі клінічних випробувань.
Білок SMN, кодований геном SMN2, неповноцінний і швидко деградує. Проте за відсутності можливості синтезу білка з мутантних алелів гена SMN1 він є єдиним джерелом білка виживання мотонейронів.
 У зв’язку зі швидкою деградацією єдина можливість підтримати рівень білка SMN в організмі – це більше продукувати його з гена SMN2. На це спрямована дія вальпроєвої кислоти та фенілбутирату. Останні в цьому випадку діють як інгібітори гістон-деацетилаз, полегшуючи зчитування інформації з ДНК. Унаслідок цього збільшується синтез білка SMN із гена SMN2 та підвищується виживання мотонейронів.
У зв’язку зі швидкою деградацією єдина можливість підтримати рівень білка SMN в організмі – це більше продукувати його з гена SMN2. На це спрямована дія вальпроєвої кислоти та фенілбутирату. Останні в цьому випадку діють як інгібітори гістон-деацетилаз, полегшуючи зчитування інформації з ДНК. Унаслідок цього збільшується синтез білка SMN із гена SMN2 та підвищується виживання мотонейронів.
У 2006 р. FDA та Європейське агентство з лікарських засобів (EMA) схвалили препарат алглюкозидаза альфа (Міозим) для лікування хвороби Помпе. Як відомо, хвороба Помпе зумовлена відсутністю чи недостатністю лізосомального ферменту кислої альфа-глюкозидази (КАГ).
Міозим містить амінокислотну послідовність і вуглеводну структуру, котрі значною мірою подібні природному попереднику КАГ у людини. Як і природний фермент, Міозим забезпечує розщеплення лізосомального глікогену до глюкози, допомогаючи запобігати розвитку інвалідизуючих ефектів накопичення глікогену. Ферментозамісна терапія алглюкозидазою альфа в пацієнтів із хворобою Помпе, по суті, заміщує те, чого не вистачає в організмі.
Мітохондріальна дисфункція супроводжує майже всі нейродегенеративні захворювання. Білки TSPO та VDAC, розташовані на зовнішній мембрані мітохондрій, вважають відповідальними за чутливість мітохондрій до вільних радикалів кисню та проникність зовнішньої мембрани. Саме вони ініціюють сигнал окисної загрози. Olesoxime – новий пероральний нейропротекторний засіб, який блокує білки TSPO та VDAC. Наразі тривають його клінічні дослідження у хворих на СМА, а також плануються випробування в пацієнтів із бічним аміотрофічним склерозом.
SRK‑016 – інактиватор міостатину, перспективний для лікування м’язової атрофії. Міостатин є ключовим сигнальним білком, який негативно впливає на м’язову масу й функцію. Також випробовується СК‑107 – активатор тропоніну (білка скорочень серцевого та скелетних м’язів). Наразі є дані про те, що СК‑107 посилює реакцію скелетних м’язів людини у відповідь на активацію нерва (J.A. Andrews et al., 2018). Проводиться його дослідження за участю хворих на СМА.
Пошуки лікування МД складніші, оскільки ген дистрофіну дуже великий і має 73 екзони. При МД можлива втрата будь-якого чи декількох із них, що зумовлює значну варіативність і складність підбору специфічної терапії. Наразі триває пошук можливостей «оминання» екзонів із мутацією. Також проводяться дослідження клітинної терапії МД шляхом уведення стовбурових попередників кардіальних клітин. Вони знижують ризик ускладнень з боку серця та сприяють підвищенню сили скелетних м’язів.
 Провідний науковий співробітник відділу дитячої психоневрології та пароксизмальних станів ДУ «Інститут неврології, психіатрії та наркології НАМН України» (м. Харків) Андрій Валерійович Шатілло представив системний погляд на проблему раннього виявлення рідкісних НМЗ на прикладі хвороби Помпе.
Провідний науковий співробітник відділу дитячої психоневрології та пароксизмальних станів ДУ «Інститут неврології, психіатрії та наркології НАМН України» (м. Харків) Андрій Валерійович Шатілло представив системний погляд на проблему раннього виявлення рідкісних НМЗ на прикладі хвороби Помпе.
З огляду на частоту захворювання, потенційна кількість хворих на хворобу Помпе в Україні – від 11 до 45 осіб у кожній області. Незважаючи на те що хвороба Помпе проявляється нескладною для клінічної оцінки симптоматикою й не потребує високотехнологічних методів діагностики на етапі скринінгу, ми не маємо стільки діагностованих випадків, очевидно, через низьку обізнаність лікарів щодо цієї патології. Тоді як своєчасний діагноз може врятувати життя хворого.
Складнощі з діагностикою й лікуванням рідкісних хвороб були, є та ще будуть якийсь час у всіх країнах світу. Європейський Союз намагається вирішувати цю проблему шляхом об’єднання в єдину мережу наявних центрів та фахівців із рідкісних і складних захворювань (European Reference Networks). Доповідач запропонував для обговорення аудиторії фахівців тему побудови подібної системи референтної діагностики в Україні із залученням Міністерства охорони здоров’я, академічних центрів, інших організацій та установ, в яких існує можливість займатися цією проблемою. Для прикладу був наведений алгоритм діагностики хвороби Помпе, який складається з трьох етапів:
1. Виявлення пацієнтів групи ризику з характерними симптомами (прогресуюча м’язова слабкість тазового та плечового поясу, швидка втомлюваність під час ходьби, труднощі під час підйому з положення сидячи/лежачи та сходами, болі в м’язах, дихальна недостатність, денна сонливість, нічна гіповентиляція, зниження маси тіла в дітей та підлітків, гіпотрофія) й лабораторними маркерами (підвищення креатинфосфокінази, амінотрансфераз, лактатдегідрогенази).
2. Специфічна діагностика: аналіз активності КАГ за методом сухої плями крові (DBS). Верифікація можлива шляхом повторного вимірювання активності КАГ у крові альтернативним методом.
3. Додаткові методи діагностики: ДНК-діагностика в разі підозри на хибно-негативний результат специфічної діагностики, біопсія м’язів.
Доповідач нагадав, що тест DBS для українських пацієнтів безкоштовно забезпечує компанія Sanofi Genzyme. Лікарям потрібно лише звернутися до представника ТОВ «Санофі-Авентіс Україна» для одержання карт для забору крові. У разі підтвердження діагнозу компанія може забезпечити хворого (за наявності симптомів, які загрожують життю) препаратом Міозим за рахунок власної гуманітарної програми до початку державних закупівель. Однак поки що, за відсутності системного підходу до проблеми за участю держави, випадки ранньої діагностики хвороби Помпе в Україні є швидше винятком, аніж належною практикою. На сьогодні в нашій країні зареєстровано всього 4 пацієнти з цією хворобою.
 Керівник Центру нервово-м’язових захворювань Львівської обласної клінічної лікарні Орест Михайлович Семеряк присвятив доповідь проблемі метаболічних міопатій.
Керівник Центру нервово-м’язових захворювань Львівської обласної клінічної лікарні Орест Михайлович Семеряк присвятив доповідь проблемі метаболічних міопатій.
Метаболічні міопатії – гетерогенна група захворювань, спільною рисою яких є порушення м’язового метаболізму, в результаті чого настає дисфункція скелетних м’язів. Більшість метаболічних міопатій зумовлені первинними генетичними ферментними дефектами, що впливають на здатність м’язових волокон підтримувати енергетичний баланс.
Метаболічні міопатії поділяють на аномалії метаболізму глікогену, ліпідів, пуринів та мітохондріальні захворювання. У групі глікогенозів виділяють до 15 типів відповідно до ферментного дефекту, а деякі хвороби мають епонімічну назву. Наприклад, глікогеноз типу 2 відомий також як хвороба Помпе.
Метаболічні міопатії рідкісні (наприклад, хвороба Помпе – 1 випадок на 40 тис. новонароджених), потенційно лікуються, їх часто помилково діагностують як м’язову дистрофію чи запальну міопатію. Міопатія дебютує зниженням толерантності до фізичного навантаження, судомами та міоглобінурією. Крампі (болючі судоми) та міалгії – патогномонічні симптоми, які можуть виникати після короткочасного фізичного навантаження (відмінна риса деяких глікогенозів, наприклад типу 5 – хвороби Мак-Ардле) чи після тривалої фізичної активності, що притаманно хворобам ліпідного обміну.
Іншими клінічними вказівками на глікогеноз можуть бути гемолітична анемія (підвищені непрямий білірубін та ретикулоцити) й затримка психічного розвитку, яка найчастіше асоціюється з дефіцитом розгалужених ферментів (тип 4) і дефіцитом альдолази А (тип 12).
При хворобі Помпе з пізнім початком пацієнти скаржаться на фіксовану проксимальну м’язову слабкість і ранню дихальну недостатність.
В інших випадках, наприклад при глікогенозі типу 3 (хвороба Корі – Форбса), дистальна м’язова слабкість може бути поєднана з кардіоміопатією та периферичною нейропатією в пацієнтів, у яких у дитинстві проявилися гепатомегалія, гіпоглікемія, котрі, як правило, покращуються з настанням пубертатного періоду.
Найпоширенішою хворобою накопичення ліпідів є дефіцит карнітинпальмітоілтрансферази. Цей білок є частиною мітохондріальної транспортної системи довголанцюгових жирних кислот, які є основним джерелом енергії для тривалої роботи скелетних м’язів. Типові гострі рецидивуючі атаки міалгії та крампі чи слабкості, часто пов’язані з міоглобінурією, можуть тривати декілька тижнів і виникати з різною частотою.
Симптоми зазвичай зумовлені тривалим фізичним навантаженням і менш часто є наслідком тривалого голодування, надмірного споживання ліпідів, охолодження, інших чинників. Патологія обмежена скелетними м’язами без ураження печінки чи серця.
При мітохондріальних захворюваннях уражаються не лише скелетні м’язи, а й центральна нервова та інші системи. Характерним є виснаження внаслідок швидкої втоми навіть під час ходьби. М’язова слабкість у спокої значно збільшується після навантаження, але, на відміну від пацієнтів із глікогенозом, не виявляється болю в м’язах, крампі чи феномену «другого дихання». Непереносимість фізичного навантаження зазвичай непропорційна відносно м’язової сили. Тому тестування сили м’язів є особливо корисним інструментом скринінгу. Також типовими є підвищений рівень лактату в спокої та виразне збільшення лактату навіть після легких фізичних вправ.
Наразі жодна з метаболічних міопатій не піддається радикальному лікуванню. Стратегія лікування включає дієтотерапію, уникання голодування, переохолодження. Ефективна ферментозамісна терапія наявна тільки для хвороби Помпе (препарат Міозим). Також для енергозабезпечення м’язів застосовується L-карнітин у дозі 60 мг/кг (вищі дози провокують міалгічний синдром). Перспективи лікування метаболічних міопатій пов’язують із генною терапією.
 Завідувач обласного медико-генетичного кабінету Закарпатської обласної клінічної лікарні ім. Андрія Новака (м. Ужгород) Еріка Йосипівна Пацкун проілюструвала складний шлях до діагнозу рідкісного захворювання на прикладі клінічного випадку хвороби Помпе в 10-річного хлопчика. Батьки звернулися до дитячого невролога, коли дитині було 9 років. Хлопчик скаржився на труднощі при ходьбі, особливо під час підйому східцями, швидку втомлюваність, болі в гомілках після навантаження. Зі слів матері, почав ходити в 1 рік і 4 міс, завжди ходив перевальцем, «був повільнішим за однолітків».
Завідувач обласного медико-генетичного кабінету Закарпатської обласної клінічної лікарні ім. Андрія Новака (м. Ужгород) Еріка Йосипівна Пацкун проілюструвала складний шлях до діагнозу рідкісного захворювання на прикладі клінічного випадку хвороби Помпе в 10-річного хлопчика. Батьки звернулися до дитячого невролога, коли дитині було 9 років. Хлопчик скаржився на труднощі при ходьбі, особливо під час підйому східцями, швидку втомлюваність, болі в гомілках після навантаження. Зі слів матері, почав ходити в 1 рік і 4 міс, завжди ходив перевальцем, «був повільнішим за однолітків».
Під час обстеження виявлено позитивний симптом Говера: «східчастий» прийом вставання з опорою руками на ноги. На електроміограмі – ознаки виразного дифузного міопатичного синдрому, більше в проксимальних м’язах. У біохімічному аналізі крові звернув на себе увагу значно підвищений рівень креатинфосфокінази – 1381 Од/л (при нормі <247 Од/л).
Дитячий невролог, до якого батьки хлопчика вперше звернулися, встановив попередній діагноз: «природжена м’язова дистрофія Дюшена». Пацієнт отримав направлення на медико-генетичну консультацію для проведення молекулярно-генетичного дослідження. ДНК-тестування на м’язову дистрофію Дюшена не виявило мутацій у 19 екзонах гена дистрофіну.
Проведено ферментне дослідження сухої плями крові на хворобу Помпе. Результат: активність альфа-глюкозидази нижча за нормальний рівень. Прицільна молекулярно-генетична діагностика виявила дві мутації, які підтвердили діагноз хвороби Помпе. Ехокардіоскопія, що проводилася при консультуванні в Центрі орфанних захворювань НДСЛ «ОХМАТДИТ», де було підтверджено діагноз, відхилень не виявила, тобто ураження міокарда в цьому випадку не відбулося. Пацієнт не має рідних братів і сестер. Планується обстеження батьків для підтвердження гетерозиготного носійства мутації. Пацієнту призначено пожиттєву ферментозамісну терапію препаратом Міозим (рекомбінантна форма КАГ людини).
Насамкінець слід наголосити, що найголовніше в діагностиці орфанних захворювань – це знання про їх існування. Лікар може зустріти один такий випадок за всю свою практику, але запідозрить патологію, лише якщо обізнаний із проблемою. Без досвідчених фахівців та ефективної системи скринінгу не мають сенсу гуманітарні ініціативи фармацевтичних компаній, які займаються розробкою терапії рідкісних захворювань. Тому надзвичайно важливе значення мають тематичні школи для лікарів та інформаційно-просвітницькі кампанії для населення. Міжнародний день хвороби Помпе, який відзначається 15 квітня, започатковано, щоби привернути увагу суспільства до потреб людей із цією рідкісною патологією.
Підготував Дмитро Молчанов
Інформація про лікарський засіб Міозим
Міозим, порошок для приготування концентрату для розчину для інфузій по 50 мг.
Склад: діюча речовина: алглюкозидаза альфа; допоміжні речовини: маніт (Е 421), натрію дигідрофосфат моногідрат, натрію гідрофосфат гептагідрат, полісорбат 80.
Показання. Тривала ферментозамісна терапія пацієнтів з підтвердженим діагнозом хвороби Помпе (дефіцит кислої α-глюкозидази). Міозим призначають дорослим та дітям будь-якого віку.
Протипоказання. Небезпечна для життя підвищена чутливість (анафілактична реакція) до активної речовини або до будь-якого з компонентів препарату.
Взаємодія з іншими лікарськими засобами та інші види взаємодій. Жодних формальних досліджень взаємодії алглюкозидази альфа з іншими лікарськими засобами не проводили.
Застосування у період вагітності або годування груддю. Вагітність. Немає даних щодо застосування алглюкозидази альфа вагітним. Період годування груддю. Алглюкозидаза альфа може проникати у грудне молоко. Фертильність. Клінічних даних щодо впливу алглюкозидази альфа на фертильність немає. Здатність впливати на швидкість реакції при керуванні автотранспортом або з іншими механізмами. Досліджень щодо здатності керувати автомобілем і користуватися технікою не проводили.
Спосіб застосування та дози. Лікування препаратом Міозим слід здійснювати під контролем кваліфікованого лікаря, який має досвід лікування пацієнтів із хворобою Помпе чи інших спадкових метаболічних або нейроваскулярних хвороб. Дози. Рекомендована схема прийому алглюкозидази альфа становить 20 мг/кг маси тіла 1 раз на 2 тижні. Передозування. Про випадки передозування не повідомлялося. У ході клінічних досліджень застосовували дози до 40 мг/кг маси тіла.
Побічні реакції. Інфантильна форма хвороби Помпе. У ході клінічних досліджень 39 пацієнтів з інфантильною формою хвороби Помпе лікували із застосуванням препарату Міозим протягом більш ніж трьох років. Побічні реакції у більшості випадків були легкого або середнього ступеня важкості, і майже всі вони виникли під час інфузії або протягом 2 годин після завершення інфузії (реакції, пов’язані з інфузією). Повідомлялося про такі серйозні реакції, пов’язані з інфузією: кропив’янка, хрипи, тахікардія, зменшення оксигенації крові, бронхоспазм, тахіпное, періорбітальний набряк та артеріальна гіпертензія.
Хвороба Помпе з пізнім початком. У ході плацебо-контрольованого дослідження, яке тривало 78 тижнів, 90 пацієнтів із формою хвороби Помпе з пізнім початком (віком від 10 до 70 років) лікували із застосуванням препарату Міозим або плацебо з рандомізацією у співвідношенні 2:1. Найчастіше спостерігалися побічні реакції, пов’язані з інфузією препарату. Більшість із цих реакцій були несерйозними, легкого або середнього ступеня важкості і зникали самостійно.
Серйозні побічні реакції, про які повідомлялося у 4 пацієнтів, яких лікували із застосуванням препарату Міозим, були наступними: ангіоневротичний набряк, відчуття дискомфорту в грудях, відчуття стиснення у горлі, некардіальний біль у грудях та суправентрикулярна (надшлуночкова) тахікардія. Реакції, які виникли у 2 з цих пацієнтів, були IgE-опосередкованими реакціями гіперчутливості.
Інформація подано скорочено. Повна інформація знаходиться в Інструкції для медичного застосування препарату Міозим, порошок для приготування концентрату для розчину для інфузій по 50 мг. Р.П. № UA/11618/01/01. Наказ МОЗ України № 1166 від 03.11.2016.
Тематичний номер «Неврологія, Психіатрія, Психотерапія» № 2 (45) червень 2018 р.