


30 серпня, 2018
Поражения глаз при системных васкулитах
Особенностью всех васкулитов является воспаление стенок сосудов [36, 70]. Глазное яблоко и окружающие его структуры богато васкуляризированы ветвями внутренней и наружной сонных артерий. При таком обширном кровоснабжении признаки васкулитов могут быть обнаружены в сосудах любого калибра.
Существуют две возможности выявления окулярной манифестации системных васкулитов (СВ): офтальмолог, который оценивает состояние глазного яблока, учитывает, может ли выявленный симптомокомплекс быть проявлением СВ; и консультация не офтальмолога – врача, чаще всего ревматолога, который диагностирует (или подозревает) СВ и пытается определить, вовлечен ли глаз в системный патологический процесс.
Поскольку глаз является единственной структурой в организме человека, в которой сосудистая сеть может быть непосредственно визуализирована, вклад офтальмолога состоит не только в сохранении зрения (так как риск потенциальной слепоты при васкулитах чрезвычайно высок), но и в диагностике, и в прогностической помощи ревматологу благодаря подтверждению или опровержению наличия и, возможно, что более важно, определению природы васкулита.
Спектр глазных проявлений при СВ широко варьирует: от достаточно безобидных эписклеритов и конъюнктивитов до тяжелых увеитов, язвенных кератитов и ишемической оптической невропатии (ИОН). Именно ИОН является одной из немногих истинных неотложных ситуаций, связанных с поражением глаза у пациентов с СВ [7].
Наиболее частым клиническим проявлением ревматических заболеваний все-таки является неинфекционный увеит [26], однако при СВ могут встречаться разнообразные поражения глаз с втягиванием в патологический процесс практически всех оболочек и структур глазного яблока.
Таким образом, все пациенты с СВ, особенно в случае наличия глазных симптомов, должны проходить комплексное обследование у специалиста-офтальмолога. А также все пациенты, консультированные окулистом, который обнаружил специфические глазные симптомы, должны быть направлены на консультацию к ревматологу для уточнения диагноза и исключения системной аутоиммунной патологии.
СВ представляют собой гетерогенную группу редких заболеваний с воспалением сосудов различного калибра в качестве общей особенности [70]. Как известно, номенклатура СВ была установлена на конференции в 1992 году [36], с недавним пересмотром на основе материалов Международной конференции Chapel Hill в 2012 году [35].
В статье представлены наиболее специфические признаки поражения оболочек глаз при различных типах СВ.
Васкулиты с поражением крупных сосудов
Преимущественно поражают аорту, ее основные ветви и соответствующие вены, хотя могут поражать и сосуды другого калибра [35].
Гигантоклеточный артериит (ГКА)
ГКА представляет собой системный некротизирующий васкулит с преимущественным поражением сосудов большого и среднего калибра, особенно это касается черепно-мозговых артерий, отходящих от сонной артерии. Приблизительно один из пяти пациентов с ГКА имеет преимущественно офтальмологические проявления заболевания [28].
Офтальмологические проявления. Для данного СВ характерны такие глазные симптомы, как потеря зрения (различной степени выраженности) в 98% случаев, преходящая слепота (31%), диплопия (6%) и боль в глазах (8%) [17]. Следует обратить внимание, что головная боль, миалгия, лихорадка более характерны для пациентов без клинических проявлений поражения глаз. Наиболее частой офтальмологической манифестацией при ГКА является передняя ИОН (80% пациентов). Это медицинское состояние, включающее потерю зрения из-за повреждения зрительного нерва по причине недостаточного кровоснабжения. При осмотре глазного дна в таких ситуациях офтальмолог диагностирует бледный отек зрительного нерва [17].
Другие васкулярные окклюзионные события, характерные для ГКА, включают окклюзию центральной ретинальной артерии (14%), окклюзию цилиоретинальной артерии (22%), заднюю ИОН (7%) [61]. Также у таких пациентов отмечают нарушения движения глазного яблока, которые могут возникнуть в результате ишемии экстраокулярных мышц [17, 70].
Вовлечение в патологический процесс обоих глаз наблюдается у одной трети пациентов с ГКА, у 5% из которых билатеральная потеря зрения остается пожизненно [24]. Постоянная потеря зрения наиболее характерна для пациентов с транзиторной потерей зрения в анамнезе, транзиторной диплопией и болями в височно-нижнечелюстном суставе [24].
Отмечено также, что низкий уровень скорости оседания эритроцитов и отсутствие симптомов системного поражения внутренних органов ассоциируются с более высоким риском развития ишемических осложнений со стороны сосудов глазного дна [59].
Точный диагноз ГКА базируется на результатах биопсии височной артерии с подтверждением наличия гранулематозного воспаления стенки артерии и разрушением муральной эластической оболочки. Традиционно биопсия проводится на стороне поражения (или на стороне с более выраженным поражением при билатеральном процессе). Breuer и соавт. оценили связь между длиной биопсии и чувствительностью диагностики.
Они обнаружили, что частота позитивных биопсий составляла 9% при длине биопсии 5 мм и меньше в сравнении с более чем 79% при длине биопсии 6-20 мм. Биопсия более 20 мм повышает уровень диагностики ГКА до 89% [12]. Эта же группа исследователей установила, что билатеральная биопсия повышает чувствительность диагностики по сравнению с унилатеральной более чем на 13% [13].
Недавние исследования по диагностике ГКА сфокусировались на альтернативной неинвазивной диагностической технике. Согласно рекомендациям Европейской противоревматической лиги (EULAR, 2018) по возможностям визуализации при васкулитах крупных сосудов в клинической практике, цветное допплер-ультразвуковое исследование височной артерии с высокой разрешающей способностью демонстрирует так называемый несжимаемый halo-сигнал концентрического гипоэхогенного утолщения стенки сосудов, что является высокоспецифичным и чувствительным признаком заболевания [18]. Кроме того, цветное допплер-ультразвуковое исследование в диагностике ГКА по специфичности можно сравнить с магнитно-резонансной томографией [18].
Артериит Такаясу (АТ)
АТ представляет собой аутоиммунный гранулематозный системный артериит, поражающий аорту и ее ветви, преимущественно у молодых женщин [54]. Васкулит вызывает деструкцию средней оболочки артерии, что приводит к развитию аневризм и реже к разрыву пораженных артерий. Гистологически он характеризуется как «панартериит», поражающий все слои сосудистой стенки, включая фиброзное утолщение интимы, деструкцию медиальных гладких мышц и эластического слоя, клеточную инфильтрацию и фиброз коллагена в средней сосудистой оболочке, наряду с утолщением адвентиции с клеточной инфильтрацией вокруг vasa vasorum [73]. Клинические симптомы зависят от уровня и степени поражения дуги аорты и отходящих от нее артерий.
Офтальмологические проявления. Именно с описания глазных симптомов, впервые сделанных японским офтальмологом Микито Такаясу и представленных на ХІІ Конгрессе Японского офтальмологического общества в 1908 году, началась история исследования неспецифического аортоартериита [1].
Ученый представил клинический случай и описание необычных изменений сосудов сетчатки (многочисленные артериовенозные фистулы), сопровождавшихся атрофией зрительного нерва, у 21-летней девушки, в анамнезе которой были синкопальные состояния.
Офтальмологическая симптоматика при АТ вызвана гипоперфузией тканей в результате облитерации сосудов. Классические признаки ретинопатии при АТ: расширение мелких сосудов, образование капиллярных микроаневризм, артерио-венозные анастомозы, гипертоническая ретинопатия, формирование таких осложнений, как катаракта, рубеоз радужки, неоваскуляризация, кровоизлияние в стекловидное тело, и другие признаки [15, 57].
Результаты крупного кросс-секционного исследования с участием пациентов с неспецифическим аортоартериитом опубликованы в 2011 году J. Peter и соавт. [57]. Согласно данным этого исследования у 61 пациента (средний возраст – 32 года; у 92% дебют заболевания приходился на возраст менее 40 лет; 77% составили женщины) были изучены офтальмологические симптомы заболевания.
Снижение зрения наблюдалось у 30% пациентов, ретинопатия Такаясу была обнаружена у 15% и гипертензивная ретинопатия – у одного из шести пациентов. Передняя ИОН была диагностирована у 3% пациентов, окулярный ишемический синдром – у 7%. Окулярный ишемический синдром чаще встречался у старших пациентов и при большей длительности заболевания. Признаки ретинопатии при АТ включали генерализованную вазодилатацию с капиллярными микроаневризмами, а также по мере развития болезни артериовенозное анастомозирование и капиллярное выпадение с последующими осложнениями, такими как катаракта, неоваскулярная глаукома, кровоизлияние в стекловидное тело и атрофия зрительного нерва.
Таким образом, 1 апреля 1908 года на XII Конгрессе Японского офтальмологического общества Микито Такаясу сделал исторический доклад по специфическим изменениям на глазном дне у женщины молодого возраста, получивший в дальнейшем всемирное признание, а описанные офтальмологом изменения вошли в симптомокомплекс одного из СВ, который теперь носит его имя.
Васкулиты с поражением сосудов среднего калибра
Сосуды среднего калибра – это висцеральные артерии и вены, а также их начальные ветви; хотя при различных подтипах васкулитов существует перекрытие в размерах сосудов, вовлеченных в воспалительный процесс [36].
Узелковый полиартериит (УПА)
УПА – это редкий некротизирующий васкулит средних или мелких артерий без гломерулонефрита или васкулита артериол, капилляров или венул. Он не связан с антинейтрофильными цитоплазматическими антителами (ANCA) и ассоциирован с аневризматическими узелками вдоль стенок мышечного слоя артерий среднего калибра [36].
Офтальмологические проявления. При УПА может поражаться любая часть тканей глазного яблока с такими проявлениями, как некроз конъюнктивы, склерит, периферический язвенный кератит (ПЯК), негранулематозный увеит, васкулит сетчатки, псевдотумор орбиты и окклюзия центральной ретинальной артерии [5]. Обычно офтальмологические проявления присутствуют у 20% пациентов с УПА [56].
Склерит, вызванный УПА, может быть диффузным, узловатым или некротизирующим. ПЯК может возникать одновременно со склеритом и прогрессировать как по окружности, так и по центру, формируя «подрытые» края, подобно язве Мурена (рис. 1); однако в отличие от язвы Мурена ПЯК при УПА часто ассоциируется со склеритом [47].
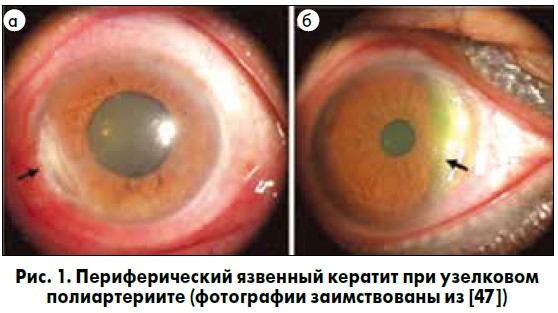
Наиболее распространенными офтальмологическими проявлениями при УПА являются хориоидальный и ретинальный васкулиты. Хориоидальный васкулит может не проявляться клинически, однако способен манифестировать в виде пятен Эльшнига (желтые пятна в сосудистой оболочке), рассеянных по всему заднему полюсу в результате хориоидальной ишемии [56, 74].
В литературе описывают следующие глазные нарушения при УПА: субгиалоидное кровоизлияние, интраретинальные геморрагии, отек, экссудаты, инфаркт фиброзного слоя нервных волокон, периваскулярные инфильтраты сосудов сетчатки, окклюзию (особенно центральной ретинальной артерии) и экссудативную отслойку сетчатки [5, 31]. При УПА также описан диффузный двусторонний панувеит (васкулит сетчатки, ассоциированный с поражением клеток передней камеры и стекловидного тела) [49].
Ткани глазницы также могут быть вовлечены в патологический процесс при УПА, что приводит к развитию орбитальной псевдотуморозной клинической картины с проптозом. Наиболее частыми нейроофтальмологическими проявлениями васкулита являются параличи черепно-мозговых нервов [23]. Отмечаются и другие проявления: гемианопсия, нистагм, транзиторная слепота, диплопия, синдром Горнера [72].
Болезнь Кавасаки (БК)
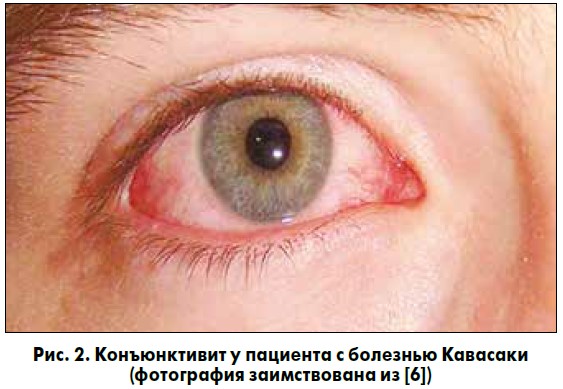 Это СВ сосудов среднего калибра, особенно коронарных артерий, возникающий преимущественно у детей в возрасте до 5 лет. Клинические проявления включают лихорадку, кожную сыпь, инъекцию конъюнктивы и цервикальную лимфаденопатию [32, 36].
Это СВ сосудов среднего калибра, особенно коронарных артерий, возникающий преимущественно у детей в возрасте до 5 лет. Клинические проявления включают лихорадку, кожную сыпь, инъекцию конъюнктивы и цервикальную лимфаденопатию [32, 36].
Офтальмологические проявления. Конъюнктивит без гнойного отделяемого является наиболее распространенным офтальмологическим проявлением БК (рис. 2), хотя не является обязательным. Также в литературе есть сведения о наличии острого переднего увеита (всегда двустороннего) [44], помутнения стекловидного тела, отека диска зрительного нерва и поверхностного точечного кератита при БК [8].
С БК также часто ассоциируется дакриоцистит, причем данное поражение манифестирует через 6 мес после начала острой фазы заболевания и является своеобразным маркером васкулита [46]. Посмертное обследование 4-месячного пациента с БК выявило васкулит с тромбозом ветви офтальмической артерии и двусторонней внутренней ишемией сетчатки [20].
Васкулиты с поражением сосудов мелкого калибра
Васкулиты, поражающие сосуды мелкого калибра, как правило, вовлекают в патологический процесс внутрипаренхимные сосуды и артерии, артериолы, капилляры, венулы и вены [36]. Данные васкулиты подразделяются на ANCA-ассоциированные и иммунокомплексные.
ANCA-ассоциированные васкулиты
Гранулематоз с полиангиитом (ГПА)
ГПА, ранее называемый гранулематозом Вегенера, – это некротизирующий гранулематозный васкулит мелких и средних сосудов с вовлечением в патологический процесс верхних и нижних дыхательных путей (в 70% случаев) [19]. Следует отметить, что вовлечение почек (развитие гломерулонефрита у 77% пациентов в развернутой стадии) и тканей глазницы также являются характерными признаками ГПА. При гистологическом исследовании тканей обнаруживают гранулематозное и негранулематозное экстраваскулярное воспаления [36].
Офтальмологические проявления. Именно поражение глаз среди патологии других органов имеет существенное клиническое и диагностическое значение при ГПА. Офтальмологические проявления возникают у 28-58% больных, с вовлечением в патологический процесс любой окулярной или периокулярной ткани, причем в 6-8% случаев – в дебюте болезни [37].
Клинически выделяют несколько фенотипов ГПА. При отсутствии поражения почек ГПА характеризуется как лимитированная форма/фенотип [43]. Однако, несмотря на существование лимитированного заболевания, ГПА редко имеет клинические проявления поражения только одного органа. Так, во французской базе данных (494 пациента с ГПА) вовлечение только одного органа наблюдалось у 16 (3,2%) больных, у 4 из которых имело место только поражение глаз. У всех этих 4 пациентов была псевдоопухоль глазницы с экзофтальмом, а у 1 – некротизирующий склерит [52].
Данные большой когорты пациентов, лечившихся в клинике, ориентированной на ГПА, в Национальных институтах здоровья США, показали, что у 15% пациентов заболевание манифестирует с поражения глаз. Наиболее распространенными дебютными симптомами были склерит и конъюнктивит, хотя заболевание глазниц с проптозом считалось наиболее диагностически значимым симптомом. У 52% пациентов в конечном итоге появлялись окулярные проявления [30].
Клиническая картина поражения глаз при ГПА может быть представлена внезапно появляющимся и быстро прогрессирующим односторонним экзофтальмом, напряженностью тканей век, их отеком; глазное яблоко оказывается «ущемленным» между напряженными веками, подвижность его резко ограничена, на роговице у лимба появляются инфильтраты и изъязвления, рано развивается отек диска зрительного нерва, и, как следствие, резко снижаются зрительные функции. Через несколько недель или месяцев аналогичная симптоматика может развиться в парной орбите.
Второй вариант течения ГПА – поражение орбиты, характеризующееся появлением невоспалительного отека век с частичным птозом и умеренным экзофтальмом на одной стороне, с постепенно развивающимся ограничением функций экстраокулярных мышц [53]. Вовлечение орбиты может возникать первично или вследствие связанного с ним синусита [4].
При отсутствии признаков поражения внутренних органов может быть установлен ошибочный диагноз – злокачественная опухоль орбиты; в таком случае диагноз может быть установлен только после диагностической орбитотомии и гистологического исследования.
Кроме поражения тканей орбиты, в патологический процесс может вовлекаться и склера. Склерит некротизирующего типа (рис. 3) является одним из распространенных офтальмологических проявлений ГПА, встречающимся у 50% пациентов [21], и может быть клинической манифестацией заболевания [29].
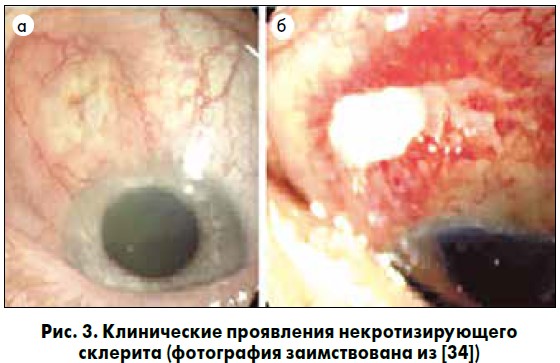
В дополнение к склериту и воспалению орбиты могут иметь место дакриоцистит, вовлечение сетчатки и роговицы [68]. Поражение слезной железы проявляется острым или хроническим дакриоаденитом, который бывает двусторонним и достаточно часто наблюдается в дебюте заболевания [66].
В патологический процесс также могут быть вовлечены такие смежные глазные структуры, как роговица, трабекулярная сетка и цилиарное тело, приводя в конечном итоге к таким серьезным осложнениям, как кератит, изъязвление роговицы, увеит, окулярная гипертензия или глаукома [9].
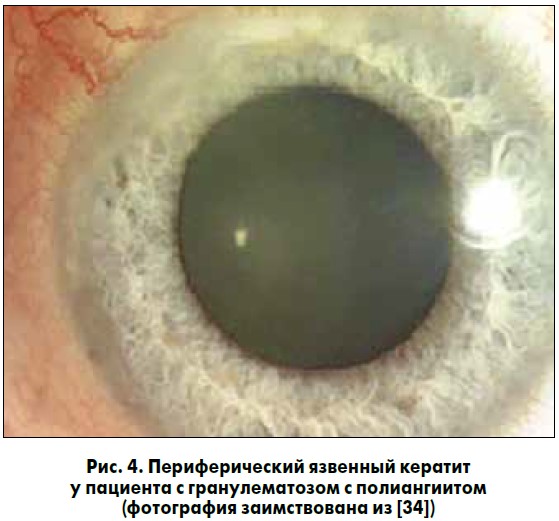 ПЯК также является распространенным окулярным проявлением ГПА (риc. 4) [21]; он часто протекает наряду со склеритом и выраженной глазной болью. При осмотре обращают на себя внимание инфильтрация и помутнение роговицы с прорастанием сосудов из лимба. Отсрочка в диагностике и лечении приводит к изъязвлению роговицы, истончению стромы и перфорации [53].
ПЯК также является распространенным окулярным проявлением ГПА (риc. 4) [21]; он часто протекает наряду со склеритом и выраженной глазной болью. При осмотре обращают на себя внимание инфильтрация и помутнение роговицы с прорастанием сосудов из лимба. Отсрочка в диагностике и лечении приводит к изъязвлению роговицы, истончению стромы и перфорации [53].
Поражение конъюнктивы может имитировать пемфигоид слизистой оболочки – развитие рубцевания конъюнктивы, потерю конъюнктивальных сводов и образование симблефарона (сращение конъюнктивы век с конъюнктивой глазного яблока) [56].
Инфильтрация при ГПА может способствовать поражению зрительного нерва, что приводит к безболезненной оптической нейропатии, отеку зрительного нерва и последующей его атрофии; дальнейшая инфильтрация может вызвать болезненную офтальмоплегию и слепоту [65]. Офтальмоплегия также может быть результатом васкулита черепных нервов, иннервирующих экстраокулярные мышцы [53].
Для подтверждения диагноза ГПА необходимо проведение биопсии пораженного органа. Классическая гистологическая диагностическая триада (васкулит мелких сосудов с инфильтрацией нейтрофилами, эозинофилами и макрофагами, очаги гранулематозного воспаления и участки некроза) при биопсии легкого обнаруживается в 90% случаев.
Диагностическая ценность гистологического исследования биоптата глазницы составляет лишь 25-54%, однако взятие биопсии из глазницы – значительно менее опасная процедура, чем биопсия почки или легкого. Сопоставления результатов биопсии глазницы с клинической картиной и лабораторными данными зачастую оказывается достаточно для постановки диагноза даже при наличии двух гистологических критериев из трех [33].
Эозинофильный ГПА (ЭГПА), или синдром Чарджа – Стросса
ЭГПА представляет собой эозинофильное некротизирующее гранулематозное воспаление с вовлечением дыхательных путей, поражающее преимущественно малые и средние сосуды. Основное клиническое проявление болезни – гиперреактивность бронхов; астма и полипы носа являются частыми клиническими симптомами, а эозинофилия – ведущая лабораторная особенность [36].
Офтальмологические проявления. ЭГПА – достаточно редкая причина заболевания глаз [62]. Описаны единичные наблюдения со множеством неспецифических проявлений, в том числе воспаление орбит, конъюнктивит, эписклерит, ПЯК, увеит, склерит, окклюзия артерий сетчатки, ИОН, мультифокальная хориоидная ишемия и параличи черепно-мозговых нервов. В дополнение к нефриту позитивность к ANCA также, по-видимому, связана с более тяжелым поражением глаз и большим риском потери зрения [67].
Микроскопический полиангиит (МПА)
Это некротизирующий васкулит мелких сосудов с возможным вовлечением средних артерий. В клинической картине МПА доминируют признаки некротизирующего гломерулонефрита и легочного капиллярита. Экстраваскулярное воспаление отсутствует [35].
Офтальмологические проявления. При МПА в воспалительный процесс могут вовлекаться все окулярные и орбитальные ткани, частота симптомов колеблется в пределах 30%. В когорте из 85 пациентов с МПА (из французской базы данных васкулитов) только у 1 пациента были отмечены окулярные проявления, но никаких подробностей о характере процесса не было представлено [27].
Наиболее распространенными формами вовлечения глаз являются ПЯК с особенностями, которые напоминают язву Мурена (зазубренный изъязвленный край), и смежный склерит [47]. Вовлечение глаз может быть начальным проявлением МПА [47]. Mihara и соавт. (2005) описали клинический случай МПА у мужчины с передним увеитом и ретинальным васкулитом [48].
Иммунокомплексные васкулиты
Криоглобулинемический васкулит (КВ)
Криоглобулинемия – наличие криоглобулинов в сыворотке, а КВ – васкулит с криоглобулинными иммунными отложениями, который поражает мелкие сосуды, преимущественно капилляры, венулы и артериолы [35].
Офтальмологические проявления. В литературе есть сведения о депозиции моноклонального иммуноглобулина IgG-k на передней пограничной мембране роговицы (боуменовой мембране) [40]. Как и в случае с другими СВ, склерит и ПЯК были отмечены у пациентов с КВ [46]. У пациентов с КВ описаны ишемия переднего сегмента с развитием неоваскуляризации радужки [39].
Сообщалось о вовлечении в процесс задних сегментов глаза при смешанной криоглобулинемии, включая Purtscher-ретинопатию, васкулит сетчатки и отслойку сетчатки, имитирующие центральную серозную хориоидопатию [11, 22].
IgA-васкулит
IgA-васкулит (васкулит Шенлейна – Геноха) характеризуется IgA1-доминантными иммунными отложениями, вовлекающими в процесс сосуды мелкого калибра, преимущественно капилляры, венулы и артериолы; в его основе лежит множественный микротромбоваскулит, поражающий сосуды кожи и внутренних органов.
При IgA-васкулите поражаются кожа, желудочно-кишечный тракт и суставы с явлениями артрита [35]. Болезнь чаще всего встречается в детском возрасте и является наиболее распространенным васкулитом детства: 3-26,7 случая на 100 тыс. детей [64] (у взрослых – 0,8-1,8 случая на 100 тыс. [58]).
Офтальмологические проявления. В литературе присутствуют сообщения о вовлечении в аутоиммунный процесс центральной нервной системы, поражение которой ведет к аномалиям зрения у 8% детей с IgA-васкулитом [33], хотя поражение глаз встречается довольно редко [64]. В научной литературе описаны случаи эписклерита, переднего увеита и кератита у пациентов с васкулитом Шенлейна – Геноха [50].
Гипокомплементемический уртикарный васкулит (ГУВ)
ГУВ (анти-C1q-васкулит) – васкулит мелких сосудов (капилляры, венулы и артериолы), встречающийся одновременно с крапивницей, гипокомплементемией и наличием анти-C1q-антител [35]. Его патогенез опосредован аутоантителами к C1q (анти-C1q) – молекулярному комплексу, входящему в состав первого компонента классического пути активации комплемента (С1) [2]. ГУВ проявляется прежде всего кожными симптомами, однако в процесс вовлекаются оболочки глаз и почечная паренхима.
Офтальмологические проявления. У 15-21% пациентов с ГУВ наблюдаются поражения глаз [16]. В литературе встречается информация о конъюнктивитах, увеитах, эписклеритах и склеритах, ассоциированных с ГУВ, что требует системной иммуносупрессии для контроля заболевания [14, 63].
Васкулиты с поражением сосудов различного калибра Болезнь Бехчета (ББ)
Это идиопатическое воспалительное заболевание, исходное описание которого включало гипопион и передний увеит как одну часть триады проявлений, оральные и половые изъязвления были двумя другими. Основополагающим в патогенезе ББ является некротизирующий васкулит с участием как артерий, так и вен [38].
Офтальмологические проявления. Заболевание глаз возникает у 70-85% пациентов с ББ. Офтальмологические проявления могут включать гипопион – патологическое скопление экссудата в передней камере глазного яблока (85-90%), передний (3,2-37%), задний (21,7-56%) или генерализованный (5,3-75,1%) увеит, васкулит сосудов сетчатки (55%) [38, 75].
Передний увеит при ББ ассоциируется с выраженной клеточной реакцией передней камеры, проявляющейся у трети больных в виде гипопиона, который отличается от такового при других воспалительных процессах тем, что состоит из лимфоцитов.
Гипопион при ББ имеет характерную подвижность: при наклоне головы пациента вперед он легко растекается по задней поверхности роговицы, а при наклоне головы назад быстро стекает на поверхность радужки. Передний увеит может быстро проходить без каких-либо последствий, однако иногда приводит ко вторичным изменениям иридо-хрусталиковой диафрагмы и угла передней камеры, осложняясь вторичной глаукомой [55].
В 95% случаев процесс поражения глаз является двусторонним, однако клинические признаки редко бывают симметричными: обычно существует определенный период между поражением глаз, который может длиться от нескольких дней до нескольких лет. Увеит обычно носит рецидивирующий характер [42]; частота рецидивов коррелирует с прогнозом заболевания.
Считается, что ББ ассоциируется с плохим прогнозом у лиц с частыми рецидивами в первый год поражения глаз. Отмечают тяжелое течение поражения глаз у пациентов с небольшим временным интервалом между началом ББ и вовлечением в процесс окулярных оболочек [71].
Кроме того, тяжесть заболевания зависит от возраста дебюта ББ: чем пациент моложе, тем хуже прогноз, особенно для поражения глаз [10]. Наиболее прогностически неблагоприятным признаком ББ является поражение заднего отрезка глаза [41]. Ангиит сетчатки еще в большей степени осложняет течение болезни, поскольку окклюзия ретинальных сосудов приводит к атрофии сетчатки и зрительного нерва [45].
Следует отметить, что главной причиной инвалидности при ББ является именно поражение глаз. Для ББ характерна постепенная облитерация сосудов сетчатки, поэтому на флюоресцентной ангиографии глазного дна отсутствует типичный для других форм ангиитов сетчатки «обрыв» ретинальных сосудов. В связи с этим при ББ редко или вообще не выявляется ангиографическая картина неперфузируемых зон и неоваскуляризации сетчатки.
Другим признаком поражения сетчатки являются ретинальные инфильтраты. Они быстро рассасываются при назначении больному глюкокортикоидов, что, скорее всего, указывает на их воспалительную природу, а не на ишемию сетчатки. Существует мнение, что эти инфильтраты являются проявлением гипопиона в сетчатке [60].
Изменения хориоидеи выявляются в значительном проценте случаев. Постоянным и непременным признаком воспаления глаза при ББ является клеточная реакция стекловидного тела, что сопровождается продукцией коллагена, приводящей к частичному или полному его фиброзу. В последнее время прогноз зрительных функций при ББ несколько улучшился благодаря ранней диагностике и применению иммуносупрессивных препаратов.
Синдром Когана (СК)
СК – это васкулит, поражающий мелкие, средние или крупные артерии (включая аорту). Это редкое аутоиммунное заболевание из группы СВ, характеризующееся воспалительным поражением глаз (интерстициальный кератит, увеит, эписклерит) и органа слуха (нейросенсорная тугоухость, вестибулярные нарушения).
Интересно, что это заболевание впервые описал в 1945 году американский офтальмолог David Cogan как сочетание неспецифического (несифилитического) интерстициального кератита и аудиовестибулярных симптомов, напоминающих болезнь Меньера [3]. Согласно последней международной классификации СВ (Chapel Hill Consensus Conference, 2012) СК относится к «васкулитам, которые поражают сосуды разного калибра» [35].
Офтальмологические проявления. СК характеризуется окулярными воспалительными поражениями; классическим проявлением болезни является язвенный интерстициальный кератит, при котором обычно прозрачная и аваскулярная строма роговицы инфильтрируется кровеносными сосудами из лимба [25].
При «атипичной» форме могут выявляться увеит, эписклерит и склерит, а интерстициальный кератит не характерен. Все формы присутствуют в ассоциации с заболеванием внутреннего уха (звон в ушах, тошнота, рвота, несколько реже атаксия) [51].
Такие поражения глаз, как папиллит, задний увеит, ретинальный васкулит и экзофтальм, обнаруживают довольно редко. У 15% больных развивается осциллопсия (иллюзия вращения окружающей обстановки) и СВ, у 10% – аортит с/без недостаточности клапанов аорты.
В Украине описан единственный случай СК у молодой женщины 25 лет, который сопровождался болями и покраснением левого глаза в дебюте болезни [3]. У пациентки отмечено развитие рецидивирующего конъюнктивита, а в последующем – левостороннего язвенного интерстициального кератита. Применение современной терапии ингибиторами фактора некроза опухоли-α позволило предотвратить развитие необратимых нарушений функции вовлеченных органов.
Выводы
СВ представляют собой гетерогенную группу редких заболеваний с воспалением сосудов различного калибра в качестве общей особенности. Глазные проявления могут быть частым симптомом при различных СВ, наиболее часто поражение глаз встречается при ГКА, ГПА, ББ и МПА [70]. Учитывая гетерогенность глазных проявлений при СВ, чрезвычайно важным является сотрудничество между ревматологами и офтальмологами, а подход к лечению этих пациентов в большинстве случаев должен быть мультидисциплинарным.
Литература
- Головач І.Ю. Історія відкриття й опису неспецифічного аортоартеріїту – хвороби Такаясу, що носить ім’я японського офтальмолога Мікіто Такаясу. Укр. ревматол. журнал, 2012; 47 (1): 97-99.
- Добронравов В.А. Гипокомплементемический уртикарный васкулит: введение в клинику и иммунобиологию. Нефрология, 2011; 15 (1): 17-26.
- Яременко О.Б., Федьков Д.Л., Шинькарук Ю.Л., Сітухо М.І., Гнілорибов А.М. Синдром Когана. Здоров’я України. Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія», 2018; 2 (57): 16-17.
- Ahmad I., Lee W.C., Nagendran V., Wilson F., Shortridge R.T.J. Localised Wegener’s granulomatosis in otolaryngology: a review of six cases. ORL J. Otorhinolaryngol. Relat. Spec. 2000; 62 (3): 149-155.
- Akova Y.A., Jabbur N.S., Foster C.S. Ocular presentation of polyarteritis nodosa. Clinical course and management with steroid and cytotoxic therapy. Ophthalmology, 1993; 100: 1775-81.
- Ball G., Fessler J., Bridges S. Oxford textbook of vasculitis. UK, Oxford university press, 2014.
- Biousse V., Newman N.J. Ischemic Optic Neuropathies. N. Engl. J. Med. 2015; 372: 2428-2436.
- Birnbaum A.D., Jiang Y., Vasaiwala R., et al. Bilateral simultaneous-onset nongranulomatous acute anterior uveitis: clinical presentation and etiology. Archives of Ophthalmology, 2012; 130: 1-6.
- Biswas J., Babu K., Gopal L., et al. Ocular manifestations of Wegener’s granulomatosis. Analysis of nine cases. Indian J. Ophthalmol. 2003; 51 (3): 217-223.
- Bonfioli A.A., Orefice F. Behcet’s disease. Semin. Ophthalmol. 2005; 20 (3): 199-206.
- Braun G.S., Horster S., Wagner K.S., et al. Cryoglobulinaemic vasculitis: classification and clinical and therapeutic aspects. Postgrad Med. J. 2007; 83 (976): 87-94.
- Breuer G., Nesher G., Nesher R. Rate of discordant findings in bilateral temporal artery biopsy to diagnose giant cell arteritis. J. Rheum. 2009; 36: 794-796.
- Breuer G., Nesher R., Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin. Exper. Rheum. 2009; 27 (Suppl. 52): S10-13.
- Buck A., Christensen J., McCarty M. Hypocomplementemic urticarial vasculitis syndrome a case report and literature review. J. Clin. Aesthet. Dermatol. 2012; 5 (1): 36-46.
- Butel N., Noel N., Touitou V., et al. Takayasu arteritis and ocular manifestations: about seven cases. ARVO journal, ARVO Annual Meeting Abstract. 2014; 55 (13): 675.
- Davis M.D., Daoud M.S., Kirby B., et al. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. Journal of the American Academy of Dermatology, 1998; 38: 899-905.
- De Smit E., O’Sullivan E., Mackey D.A., Hewitt A.W. Giant cell arteritis: ophthalmic manifestations of a systemic disease. Graefes Archiv fur Ophthalmologie, 2016; 254 (12): 2291-2306.
- Dejaco C., Ramiro S., Duftner C., et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann. Rheum. Dis. 2018; 77 (5): 636-643.
- Falk R.J., Gross W.L., Guillevin L., et al. American College of Rheumatology, American Society of Nephrology and European League Against Rheumatism. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011; 63: 863-864.
- Font R.L., Mehta R.S., Streusand S.B., et al. Bilateral retinal ischemia in Kawasaki disease. Postmortem findings and electron microscopic observations. Ophthalmology, 1983; 90: 569-77.
- Galor A., Thorne J.E. Scleritis and peripheral ulcerative keratitis. Rheumatic Diseases Clinics of North America, 2007; 33: 835-854.
- Garrity J. Ocular manifestations of small-vessel vasculitis. Cleveland Clinic Journal of Medicine, 2012; 79 (3): S31-S33.
- Golnik K.C. Neuro-ophthalmologic manifestations of systemic disease: rheumatologic/Inflammatory. Ophthalmology Clinics of North America, 2004; 17: 389-96.
- Gonzalez-Gay M.A., Martinez-Dubois C., Agudo M., et al. Giant cell arteritis: epidemiology, diagnosis, and management. Current Rheumatology Reports, 2010; 12: 436-42.
- Grasland A., Pouchot J., Hachulla E., et al. Study Group for Cogan’s syndrome. Typical and atypical Cogan’s syndrome: 32 cases and review of the literature. Rhumatology (Oxford), 2004; 43: 1007-15.
- Gritz D.C., Wong I.G. Incidence and prevalence of uveitis in northern California: The northern California epidemiology of uveitis study. Ophthalmology, 2004; 111: 491-500.
- Guillevin L., Durand-Gasselin B., Cevallos R., et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999; 42: 421-30.
- Hayreh S.S., Podhajsky P.A., Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am. J. Ophthal. 1998; 125: 521-6.
- Hijikata N., Takayanagi N., Yoneda K., et al. A case of scleritis as the initial clinical manifestation of limited Wegener’s granulomatosis. Nihon Kokyuki Gakkai Zasshi. 2009; 47 (11): 1025-1029.
- Hoffman G.S., Kerr G.S., Leavitt, R.Y., et al. Wegener granulomatosis: an analysis of 158 patients. Ann. Inter. Med. 1992; 116: 488-98.
- Hsu C.T., Kerrison J.B., Miller N.R., Goldberg M.F. Choroidal infarction, anterior ischemic optic neuropathy, and central retinal artery occlusion from polyarteritis nodosa. Retina, 2001; 21: 348-51.
- Iannetti L., Zito R., Bruschi S., et al. Recent understanding on diagnosis and management of central nervous system vasculitis in children. Clinical and Developmental Immunology, 2012; 698327.
- Isa H., Lightman S., Luthert P.J., et al. Histopathological features predictive of a clinical diagnosis of ophthalmic granulomatosis with polyangiitis (GPA). Int. J. Clin. Exp. Pathol. 2012; 5 (7): 684-689.
- Isa H., Lightman S., Pusey D., Taylor S. Ocular manifestations of Wegener’s granulomatosis. J. Expert Rev. Ophthal. 2011; 6 (5): 541-555.
- Jennette J., Falk R., Bacon P., et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013; 65: 1-11.
- Jennette J.C., Falk R.J., Andrassy K., et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994; 37: 187-92.
- Joshi L., Hamour S., Salama A.D., et al. Renal and ocular targets for therapy in Wegener’s granulomatosis. Inflamm. Allergy Drug Targets. 2009; 8 (1): 70-79.
- Kacmaz R.O., Kempen J.H., Newcomb C., et al. Systemic Immunosuppressive Therapy For Eye Diseases Cohort Study Group Ocular inflammation in Behcet disease: incidence of ocular complications and of loss of visual acuity. Am. J. Ophthal. 2008; 146: 828-836.
- Kedhar S.R., Belair M.L., Jun A.S., et al. Scleritis and peripheral ulcerative keratitis with hepatitis C virus-related cryoglobulinemia. Arch. Ophthal. 2007; 125: 852-853.
- Kremer I., Wright P., Merin S., et al. Corneal subepithelial monoclonal kappa IgG deposits in essential cryoglobulinaemia. Br. J. Ophthal. 1989; 73: 669-673.
- Kump L.I., Moeller K.L., Reed G.F., et al. Behcet’s disease: comparing 3 decades of treatment response at the National Eye Institute. Can. J. Ophthalmol. 2008; 43 (4): 468-472.
- Kural-Seyahi E., Fresko I., Seyahi N., et al. The long-term mortality and morbidity of Behcet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore), 2003; 82 (1): 60-76.
- Lamprecht P., Gross W.L. A brief history of Wegener’s granulomatosis: on limited, localized, and generalized forms of the disease: comment on the article by the Wegener’s Granulomatosis Etanercept Trial Research Group. Arthritis Rheum. 2004; 50: 334-335.
- Lee K.J., Kim H.J., Kim M.J., et al. Usefulness of anterior uveitis
- as an additional tool for diagnosing incomplete Kawasaki disease. Korean J. Pediatr. 2016; 59 (4): 174-177.
- Matsuo T., Itami M., Nakagawa H., Nagayama M. The incidence and pathology of conjunctival ulceration in Behcet’s syndrome. Br. J. Ophtalmol. 2002; 86 (2): 140-3.
- Mauriello J.A. Jr., Stabile C., Wagner R.S. Dacryocystitis following Kawasaki’s disease. Ophthalmic Plastic and Reconstructive Surgery, 1986; 2: 209-211.
- Messmer E.M., Foster C.S. Vasculitic peripheral ulcerative keratitis. Survey of Ophthalmology, 1999; 43: 379-396.
- Mihara M., Hayasaka S., Watanabe K., et al. Ocular manifestations in patients with microscopic polyangiitis. Eur. J. Оphthal. 2005; 15: 138-142.
- Morgan C.M., Foster C.S., D’Amico D.J., Gragoudas E.S. Retinal vasculitis in polyarteritis nodosa. Retina, 1986; 6: 205-9.
- Muqit M., Gallagher M., Gavin M., et al. Henoch-Schonlein purpura with keratitis and granulomatous anterior uveitis. Br. J. Ophthalmol. 2005; 89 (9): 1221-1222.
- Pagnini I., Zannin M.E., Vittadello F., et al. Clinical features and outcome of Cogan syndrome. Journal of Pediatrics, 2012; 160: 303-307e1.
- Pagnoux C., Stubbe M., Lifermann F., et al. Wegener’s granulomatosis strictly and persistently localized to one organ is rare: assessment of 16 patients from the French Vasculitis Study Group database. J. Rheumatol. 2011; 38: 475-8.
- Pakrou N., Selva D., Leibovitch I. Wegener’s granulomatosis: ophthalmic manifestations and management. Semin. Arthritis Rheum. 2006; 35 (5): 284-292.
- Panja M., Mondal P.C. Current status of aortoarteritis in India. Journal of the Association of Physicians of India, 2004; 52: 48-52.
- Paovic J., Paovic P., Sredovic V. Behcet’s Disease: Systemic and Ocular Manifestations. Biomed. Res. Int. 2013; Article ID: 247345.
- Perez V.L., Chavala S.H., Ahmed M., et al. Ocular manifestations and concepts of systemic vasculitides. Survey of Ophthalmology, 2004; 49: 399-418.
- Peter J., David S., Danda D., et al. Ocular manifestations of Takayasu arteritis. Retina, 2011; 31: 1170-8.
- Piram M., Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch–Schonlein): current state of knowledge. Curr. Opin. Rheumatol. 2013; 25 (2): 171-8.
- Ponte C., Rodrigues A., O’Neill L., Luqmani R. Giant cell arteritis: Current treatment and management. World J. Clin. Cases, 2015; 3 (6): 484-494.
- Ramsay A., Lightman S. Hypopyon uveitis. Surv. Ophthalmol. 2001; 46 (1): 1-18.
- Rizzo I.V., Joseph F. Ophthalmic manifestations of giant cell arteritis.
- III Rheumatology, 2018; 57 (2-1): ii63-i72.
- Rosenbaum J.T., Ku J., Ali A., et al. Patients with retinal vasculitis rarely suffer from systemic vasculitis. Sem. Arthritis Rheum. 2012; 41: 859-65.
- Roy K., Talukdar A., Kumar B., Sarkar S. Case Report Hypocomplementaemic urticarial vasculitis syndrome: a mimicker of systemic lupus erythematosus. BMJ Case Rep. 2013; bcr2013009082.
- Saulsbury F.T. Henoch–Schonlein purpura. Curr. Opin. Rheumatol. 2001; 13: 35-40.
- Shunmugam M., Morley A.M.S., Graham E., et al. Primary Wegener’s granulomatosis of the orbital apex with initial optic nerve infiltration. Orbit. 2011; 30 (1): 24-26.
- Soheilian M., Bagheri A., Aletaha M. Dacryoadenitis as the earliest presenting manifestation of systemic Wegener’s granulomatosis. Eur. J. Ophthalmol. 2002; 12 (3): 241-243.
- Takanashi T., Uchida S., Arita M., et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg–Strauss syndrome: report of two cases and review of the literature. Ophthalmology, 2001; 108: 1129-33.
- Tarabishy A.B., Schulte M., Papaliodis G.N., Hoffman G.S. Wegener’s granulomatosis: clinical manifestations, differential diagnosis, and management of ocular and systemic disease. Surv. Ophthalmol. 2010; 55 (5): 429-444.
- Telander D.G., Holland G.N., Wax M.B., Van Gelder R.N. Rubeosis and anterior segment ischemia associated with systemic cryoglobulinemia. Am. J. Ophthal. 2006; 142: 689-90.
- Tugal-Tutkun I. Systemic vasculitis and the eye. Curr. Opin. Rheumatol. 2017; 29 (1): 24-32.
- Tugal-Tutkun I., Onal S., Altan-Yaycioglu R., et al. Uveitis in Behcet disease: an analysis of 880 patients. Am. J. Ophthal. 2004; 138: 373-80.
- Ueno K., Matsushima A., Hineno A., et al. Polyarteritis nodosa with central nervous system involvement mimicking relapsing-remitting multiple sclerosis. Mod. Rheumatol. 2014; 24 (3): 525-8.
- Vaideeswar P., Deshpande J. Pathology of Takayasu arteritis: A brief review. Ann. Pediatr. Cardiol. 2013; 6 (1): 52-58.
- Vazquez-Romo K., Rodriguez-Hernandez A., Paczka J., et al. Ocular presentation of polyarteritis nodosa. Clinical course and management with steroid and cytotoxic therapy. Front Neurol. 2017; 8: 490.
- Zamir E., Bodaghi B., Tugal-Tutkun I., et al. Conjunctival ulcers in Behcet’s disease. Ophthalmology, 2003; 110: 1137-41.
Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» № 3 (58) червень 2018 р.