


27 вересня, 2018
Ідіопатичний фіброз легень: що потрібно знати лікарю первинної ланки
Ідіопатичний фіброз легень (ІФЛ) є важким та потенційно смертельним захворюванням, що переважно уражає осіб похилого віку. ІФЛ характеризується рентгенографічною та гістопатологічною картиною звичайної інтерстиційної пневмонії (ЗІП) без жодних відомих причин розвитку.
Для ведення пацієнтів з ІФЛ необхідний точний діагноз, тому автори рекомендують якомога швидше скеровувати таких хворих до спеціалізованих центрів інтерстиційних хвороб легень для підтвердження діагнозу, початку відповідного лікування, пояснення пацієнту прогнозу захворювання, залучення до участі в реєстрах хвороби та клінічних дослідженнях, підбору кандидатів для трансплантації легень. Лікарям первинної ланки часто трапляються пацієнти з ІФЛ при зверненні хворого з відповідними скаргами чи при випадковому виявленні характерних змін під час проведення комп’ютерної томографії (КТ).
Що таке ІФЛ?
ІФЛ – це одна з більш ніж 150 інтерстиційних хвороб легень, які об’єднані неспецифічними симптомами на кшталт задишки та сухого кашлю з поступовим початком, певними рентгенологічними змінами та розвитком рестриктивної дихальної недостатності [1]. ІФЛ можна розподілити на різні ідіопатичні інтерстиційні пневмонії: ідіопатичну неспецифічну інтерстиційну пневмонію, респіраторну бронхіоліт-асоційовану інтерстиційну хворобу легень, дифузний альвеолярний крововилив, криптогенну організуючу пневмонію, гостру інтерстиційну пневмонію [2].
Для встановлення діагнозу ІФЛ слід виявити ЗІП та виключити інші причини інтерстиційних хвороб легень (захворювання сполучної тканини, реакція на лікування, інгаляція певних речовин, пневмоконіози, інфекційний та неінфекційний гранулематози).
Справжню поширеність захворюваності на ІФЛ важко оцінити. Загалом, ІФЛ вважається рідкісною хворобою, але насправді він зустрічається частіше, ніж вважалося раніше. У 2011 р. Raghu і співавт. [3] оцінили розповсюдженість ІФЛ серед користувачів системи Medicare як 495 випадків на 100 тис. Відповідно до цієї оцінки, у США може бути до 160 тис. хворих на ІФЛ [4]. Raghu і співавт. також продемонстрували, що ІФЛ частіше уражає осіб від 65 років, а це свідчить, що у процесі старіння популяції частота цього захворювання може зростати [5].
Крім того, у зв’язку зі збільшенням багаторазового застосування КТ для скринінгу раку легень діагностуватиметься все більше випадків ІФЛ [6-8]. Відповідно до старих даних, затримка від початку симптомів до визначення точного діагнозу становила 1-2 роки [9]. Більш нове дослідження виявило, що затримка у скеруванні пацієнтів з ІФЛ до медичних закладів третинного рівня асоціюється з більшим рівнем смертності незалежно від тяжкості хвороби [10].
ІФЛ – зазвичай прогресуюче і часто фатальне захворювання
Здебільшого ІФЛ являє собою захворювання, що прогресує, обмежене легенями та характеризується поганим прогнозом. Медіана виживання становить приблизно 2-5 років від постановки діагнозу, хоча ця оцінка ґрунтується на старих даних, які не відображають теперішніх інновацій у діагностиці та лікуванні. Недавні дослідження свідчать про більшу частку виживання у разі збереження функції легень [11].
Як свідчить назва хвороби, етіологія ІФЛ невідома, але наукові роботи вказують на вагому роль генетичної схильності в більшості випадків [12]. Незалежно від причини, патогенез та прогресування ІФЛ вважаються результатом аномальної стійкої ранозагоювальної відповіді. Відкладення рубцевої тканини, що прогресує, порушує нормальну архітектуру та функцію легень, зрештою спричиняючи клінічно виражене захворювання [13].
Симптоматика
ІФЛ зазвичай розпочинається з поступового наростання задишки при фізичному навантаженні з хронічним кашлем чи без нього. Фактори ризику включають чоловічу стать, похилий вік, анамнез куріння. Пацієнти з недіагностованим ІФЛ, які звертаються до лікаря з задишкою та повідомляють про куріння в анамнезі, переважно підлягають емпіричному лікуванню з приводу хронічних обструктивних захворювань легень.
Типовою ознакою ІФЛ при аускультації є вологі хрипи, що може вплинути на призначення надмірної кількості діагностичних процедур, спрямованих на серцево-судинну систему, та емпіричного лікування з приводу серцевої недостатності (СН). Порівняно часто трапляється деформація пальців у вигляді барабанних паличок [14]. Іншою типовою рисою ІФЛ є гіпоксемія при фізичному навантаженні, яка часто корелює з важкістю хвороби та її прогнозом. На пізніших стадіях відзначається гіпоксемія у спокої.
Під час спірометрії у пацієнтів з ІФЛ зазвичай виявляються рестриктивні зміни, тобто нормальне чи збільшене співвідношення об’єму форсованого видиху за 1-шу секунду до форсованої життєвої ємності легень (ОФВ1/ФЖЄЛ) (>70% чи більше нижньої межі норми) у поєднанні зі зниженою ФЖЄЛ. Про рестриктивні зміни також свідчить знижена загальна ємність легень (<80% від передбачуваного рівня чи менше нижньої межі норми) при проведенні плетизмографії. Часто зустрічається порушений газообмін, що проявляється зниженою дифузійною ємністю легень для монооксиду вуглецю (ДЄМВ). Оскільки перфузія легень вища у базальних ділянках, де частіше виникає ІФЛ, ДЄМВ часто більш виражено знижена, ніж ФЖЄЛ.
Прогностичні фактори
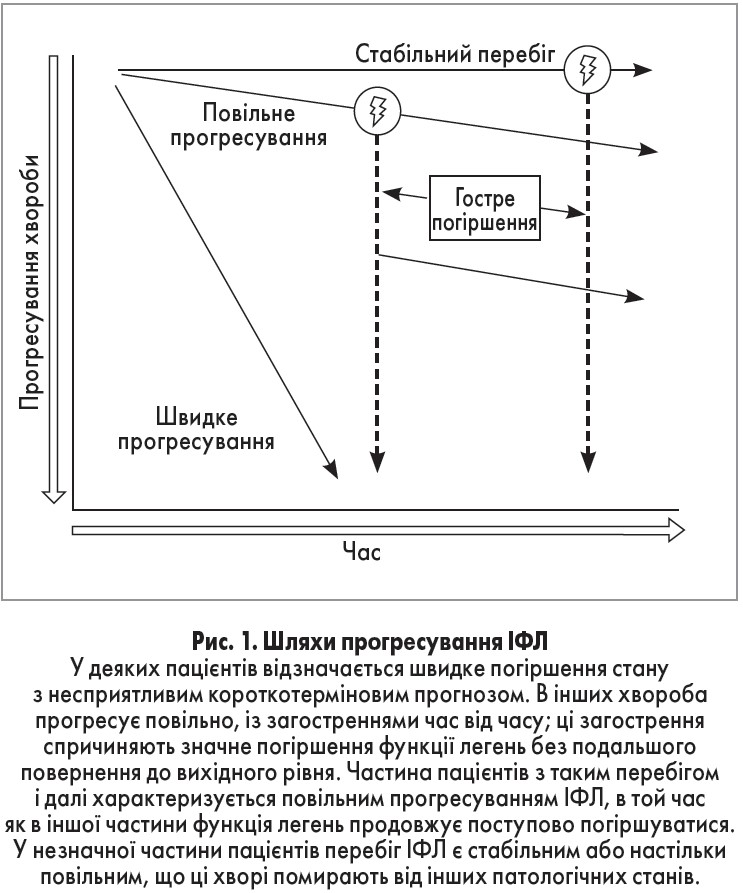 Клініцисти зазвичай розглядають ІФЛ як невпинний прогресуючий процес, але перебіг цієї хвороби є варіабельним і може відрізнятися у різних пацієнтів (рис. 1) [15,16]. Проте з часом у більшості пацієнтів розвивається зниження функції легень, що веде до дихальної недостатності. Дихальна недостатність, якій зазвичай передує підгостре погіршення (триває кілька тижнів – кілька місяців) чи гостре погіршення (триває <4 тиж), є найчастішою причиною смерті за наявності ІФЛ, однак коморбідні хвороби (рак легень, інфекційні процеси, серцева недостатність) у таких пацієнтів також часто завершуються фатально [17, 18].
Клініцисти зазвичай розглядають ІФЛ як невпинний прогресуючий процес, але перебіг цієї хвороби є варіабельним і може відрізнятися у різних пацієнтів (рис. 1) [15,16]. Проте з часом у більшості пацієнтів розвивається зниження функції легень, що веде до дихальної недостатності. Дихальна недостатність, якій зазвичай передує підгостре погіршення (триває кілька тижнів – кілька місяців) чи гостре погіршення (триває <4 тиж), є найчастішою причиною смерті за наявності ІФЛ, однак коморбідні хвороби (рак легень, інфекційні процеси, серцева недостатність) у таких пацієнтів також часто завершуються фатально [17, 18].
Предикторами смертності є зниження ФЖЄЛ та ДЄМВ, посилення симптомів та фізіологічних порушень, що проявляється зниженням відстані у тесті 6-хвилинної ходьби чи посиленням гіпоксемії при фізичному навантаженні [19-22]. Іншими частими коморбідними станами, пов’язаними з погіршенням якості життя та поганим прогнозом, є обструктивне апное сну, гастроезофагальна рефлюксна хвороба, депресія [16, 23].
Ретроспективні дослідження свідчать, що більшість пацієнтів з ІФЛ помирають впродовж 2-5 років після початку симптомів. Враховуючи затримку між початком симптомів та встановленням остаточного діагнозу, середня очікувана тривалість життя після діагностики ІФЛ спостерігається в межах 2 років [9, 18, 24, 25].
Для прогнозування ризику коротко- та довготермінової смертності були створені дві системи розподілу на стадії ІФЛ, що ґрунтуються на статі, віку та фізіологічних параметрах [23, 24]. Зокрема, індекс GAP (gender – стать, age – вік, physiology – фізіологія) забезпечує оцінку ризику смерті за шкалою від 0 до 8 балів. Надалі бальна оцінка пацієнта категоризується як стадії I, II та III. Для кожної стадії встановлені приблизні показники 1-, 2- та 3-річної смертності. Найвищими ці показники є для III стадії.
Калькулятор GAP (www.acponline.org/journals/annals/extras/gap) дозволяє розрахувати приблизний ризик для кожного окремого пацієнта. Застосування подібних калькуляторів у веденні пацієнтів з ІФЛ лише впроваджується, однак вони можуть бути корисними для пояснення пацієнту прогнозу хвороби.
Діагностика
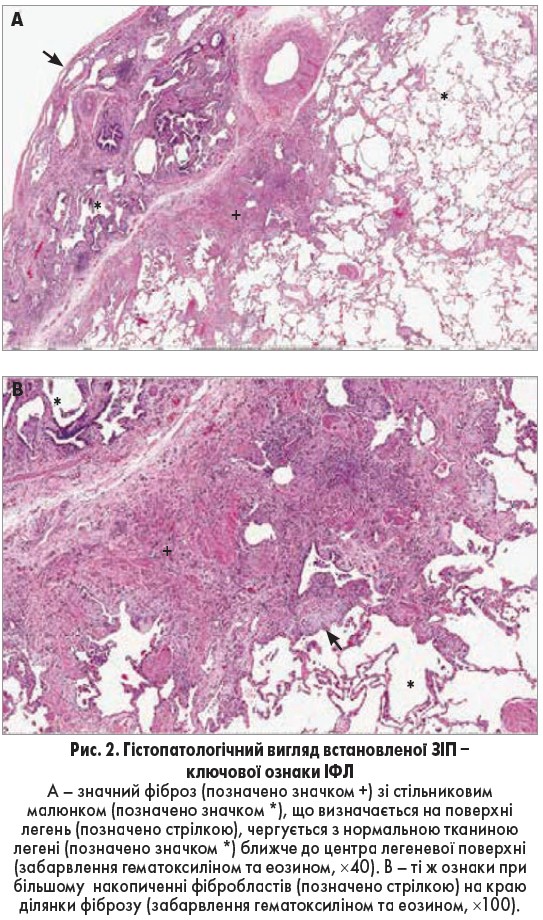 ЗІП супроводжує специфічна гістологічна картина, яку можна побачити на зразках тканини легень, узятих при хірургічній біопсії. Ця картина характеризується фіброзом та малюнком, який нагадує стільники, що чергуються з ділянками нормальної легеневої паренхіми (рис. 2). Ці порушення зазвичай розміщені субплеврально та більш виражені у нижніх частках. Запалення у переважній більшості випадків не визначається.
ЗІП супроводжує специфічна гістологічна картина, яку можна побачити на зразках тканини легень, узятих при хірургічній біопсії. Ця картина характеризується фіброзом та малюнком, який нагадує стільники, що чергуються з ділянками нормальної легеневої паренхіми (рис. 2). Ці порушення зазвичай розміщені субплеврально та більш виражені у нижніх частках. Запалення у переважній більшості випадків не визначається.
ЗІП можна виявити при гістологічному дослідженні зразків легень не тільки за умов ІФЛ, а й при інших фібротизуючих хворобах легень, у т. ч. при інтерстиційних хворобах легень, асоційованих із захворюваннями сполучної тканини чи утворених внаслідок інгаляційних чи професійних пошкоджень; при хронічному гіперчутливому пневмоніті [26-29]. Отже, постановка діагнозу ІФЛ вимагає виключення інших відомих причин ЗІП.
Відповідно до рекомендацій 2011 р. [16], гістологічна картина інтерстиційного захворювання легень дозволяє точно діагностувати чи запідозрити ЗІП, а також виявити атипову картину, що свідчить про інший діагноз.
У номенклатурі ЗІП описана також картина хвороби при проведенні КТ з високою роздільною здатністю (КТВРЗ). КТВРЗ проводиться без контрасту та дозволяє отримати зображення тонких зрізів (зазвичай <1,5 мм) при вдиху, видиху та у положенні долілиць, що забезпечує виявлення своєрідних повітряних пасток, наявність яких може свідчити про інший діагноз, пов’язаний з ураженням дихальних шляхів, а не паренхіми легень.
На зображеннях КТВРЗ ЗІП виглядає як сітчасті затемнення, часто з тракційними бронхоектазами чи бронхіолектазами, які зазвичай розташовані у базальних ділянках та на периферії легень. Ключовою ознакою є стільниковий малюнок, який виглядає як скупчення кіст з добре вираженими стінками на периферії легеневої паренхіми. Затемнення на зразок матового скла не є типовою ознакою ЗІП, і, хоча вони не виключають такого діагнозу, за їх наявності треба розглянути ймовірність інших патологічних станів [16]. Ще однією поширеною ознакою ЗІП є реактивна аденопатія лімфовузлів середостіння та воріт легень.
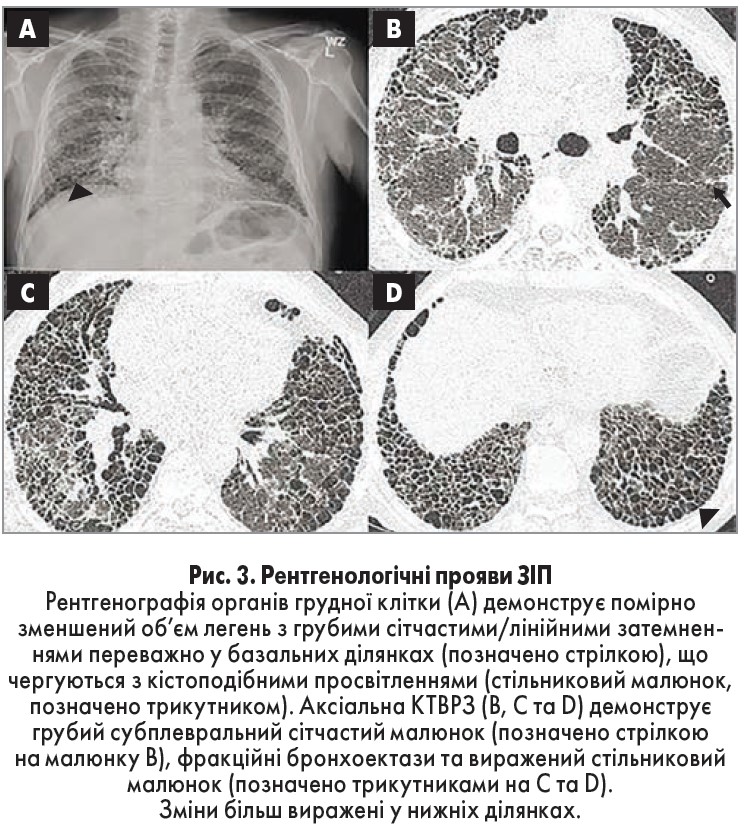 При оцінці результатів КТВРЗ рентгенолог категоризує отриману картину як точно визначену ЗІП, ймовірну ЗІП чи непевний результат. Точно визначена ЗІП відповідає усім перерахованим вище критеріям та не має жодних ознак, що можуть свідчити про альтернативний діагноз (рис. 3).
При оцінці результатів КТВРЗ рентгенолог категоризує отриману картину як точно визначену ЗІП, ймовірну ЗІП чи непевний результат. Точно визначена ЗІП відповідає усім перерахованим вище критеріям та не має жодних ознак, що можуть свідчити про альтернативний діагноз (рис. 3).
Ймовірна ЗІП має всі перераховані вище ознаки, окрім стільникового малюнка. Якщо отриману при КТВРЗ картину схарактеризовано як точно визначену ЗІП, біопсія не обов’язкова, але якщо ж результат непевний чи виявлено ймовірну ЗІП для постановки діагнозу ІФЛ необхідна хірургічна біопсія.
Однак з’являється все більше доказів, що за умов відповідного клінічного сценарію ймовірна ЗІП перебігає аналогічно точно визначеній ЗІП, тому картина ймовірної інтерстиційної пневмонії є достатньою для постановки клінічного діагнозу ІФЛ навіть без підтвердження за допомогою біопсії [30].
Діагностичний алгоритм для ІФЛ
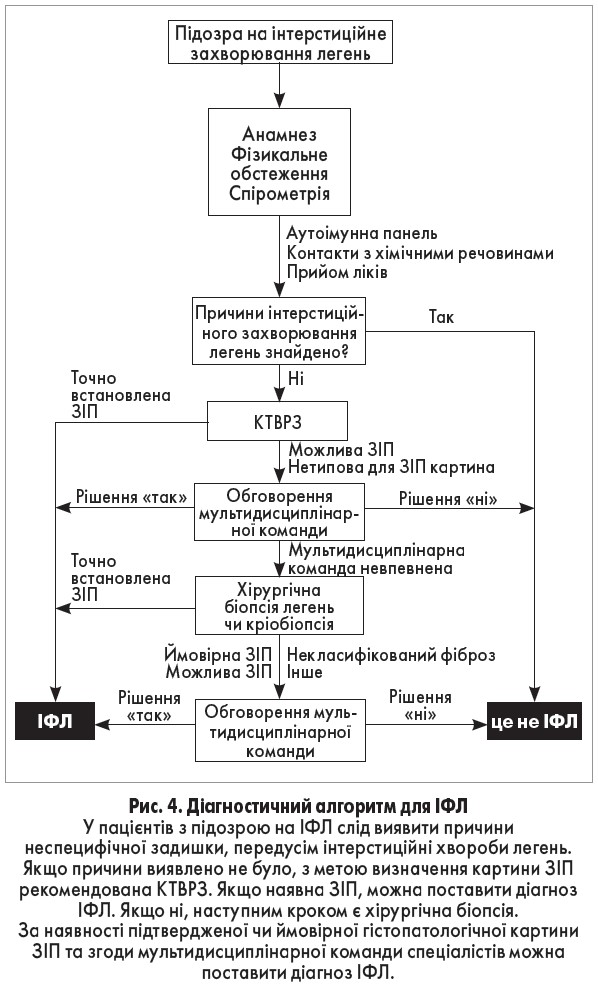
Враховуючи велику кількість інтерстиційних хвороб легень, їх складність та відсутність золотого стандарту діагностичних методів, діагностика ІФЛ (табл.) може бути досить складною і вимагати інтеграції клінічних, рентгенологічних та, за потреби, патологоанатомічних характеристик.
 Загальною картиною ЗІП при візуалізаційних дослідженнях чи біопсії легень можуть бути множинні патологічні процеси, які необхідно виключити до постановки діагнозу ІФЛ. Для чого Американське торакальне товариство, Європейське респіраторне товариство, Японське респіраторне товариство та Латиноамериканська торакальна асоціація сформулювали об’єднані рекомендації з діагностики та лікування ІФЛ [16]. Автори пропонують оновлений алгоритм, який дещо відрізняється від цих рекомендацій (рис. 4).
Загальною картиною ЗІП при візуалізаційних дослідженнях чи біопсії легень можуть бути множинні патологічні процеси, які необхідно виключити до постановки діагнозу ІФЛ. Для чого Американське торакальне товариство, Європейське респіраторне товариство, Японське респіраторне товариство та Латиноамериканська торакальна асоціація сформулювали об’єднані рекомендації з діагностики та лікування ІФЛ [16]. Автори пропонують оновлений алгоритм, який дещо відрізняється від цих рекомендацій (рис. 4).
Демографічні характеристики та фактори ризику пацієнта повинні звернути увагу лікаря на можливість наявності ІФЛ. Ймовірність ІФЛ наростає з віком, і частіше ця хвороба зустрічається у чоловіків. Ще одним фактором ризику є куріння в анамнезі [31]. Отже, 45-річний чоловік, який ніколи не курив, менш імовірно має ІФЛ, ніж 70-річний колишній курець, а 70-річний чоловік більш імовірно має ІФЛ, ніж жінка того ж віку. Отже, виявлення інтерстиційної хвороби легень в особи з нетиповим демографічним профілем (наприклад молода жінка, яка ніколи не курила) має стати приводом для ретельного пошуку іншого діагнозу, зокрема гіперчутливого пневмоніту чи захворювань сполучної тканини.
Після врахування демографічного профілю та факторів ризику слід точно та ретельно зібрати анамнез, який має містити в собі оцінку тяжкості задишки та кашлю, виявлення ознак та симптомів хвороб сполучної тканини (артралгії, симптоми сухості шкіри, слизових оболонок, очей, феномен Рейно, труднощі при ковтанні), а також гастроезофагальної рефлюксної хвороби, яка може асоціюватися з хворобами сполучної тканини та з ІФЛ.
Важливо також ідентифікувати будь-які чинники зовнішнього середовища, які можуть викликати пневмоконіоз чи хронічний гіперчутливий пневмоніт. Найбільш розповсюдженими факторами ризику останнього є пташине пір’я, пліснява, грибок, застосування гарячих ванн, деякі промислові хімікати [32].
Важливим є анамнез приймання лікарських засобів. З інтерстиційними хворобами легень можуть асоціюватися багато медикаментів, але аміодарон, блеоміцин, метотрексат та нітрофурантоїн викликають подібні стани найчастіше [33]. Оскільки існують сімейні форми ІФЛ, необхідно уважно зібрати сімейний анамнез.
 Фізикальне обстеження повинне включати уважну аускультацію з метою пошуку хрипів, хоча ця ознака не специфічна для ІФЛ, а є найбільш поширеним відхиленням під час вислуховування легень. Детальний огляд шкіри, обстеження м’язово-скелетної та серцево-судинної систем є важливими для виявлення ознак ревматологічних хвороб, деформації пальців, доказів СН чи легеневої гіпертензії.
Фізикальне обстеження повинне включати уважну аускультацію з метою пошуку хрипів, хоча ця ознака не специфічна для ІФЛ, а є найбільш поширеним відхиленням під час вислуховування легень. Детальний огляд шкіри, обстеження м’язово-скелетної та серцево-судинної систем є важливими для виявлення ознак ревматологічних хвороб, деформації пальців, доказів СН чи легеневої гіпертензії.
Лабораторні обстеження мають включати серологічну панель антитіл для виявлення хвороб сполучної тканини, які можуть проявитися інтерстиційним захворюванням легень, у т. ч. ревматоїдного артриту, дерматополіміозиту, склеродермії, синдрому Шегрена, інших недиференційованих чи поєднаних патологічних станів.
Типові лабораторні тести містять антинуклеарні антитіла, ревматоїдний фактор, визначення швидкості осідання еритроцитів, С-реактивного протеїну. Крім того, можуть призначатися дослідження на антициклічний цитрулінований пептид (анти-CCP), анти-Scl‑70, анти-RNP, анти-SS-A, анти-SS- B та анти-Jo‑1 [16]. Обсяг панелі має залежати від демографічних характеристик пацієнта, даних анамнезу та фізикального обстеження, які збільшують чи зменшують імовірність виявлення хвороб сполучної тканини.
Оцінка функції легень пацієнта має складатися зі спірометрії, ДЄМВ, плетизмографії. Здебільшого ІФЛ супроводжується рестриктивними змінами. Оскільки рестриктивні зміни характеризуються низькою загальною ємністю легень, визначення ФЖЄЛ може бути сурогатним показником як дешевший та легший у виконанні метод. Загалом, нижча загальна ємність легень чи ФЖЄЛ є показниками важчого ураження. ДЄМВ є ще одним маркером тяжкості захворювання, але менш надійним у зв’язку з більшою варіабельністю та складнощами у проведенні необхідного для обстеження маневру.
Тест з 6-хвилинною ходьбою також є важливим інструментом оцінки фізіологічних функцій, який дозволяє визначити гіпоксемію при фізичному навантаженні та функціональний статус хворого, що у подальшому допомагає у з’ясуванні прогнозу.
У процесі діагностичного пошуку більшість пацієнтів з задишкою нез’ясованого генезу підлягають рентгенографії органів грудної клітки. Однак це обстеження не дозволяє сформулювати точний діагноз за підозри на інтерстиційне захворювання легень і не може виключити зміни, що відображають ранні фази захворювання. У міру прогресування хвороби звичайна рентгенограма може виявити сітчасто-вузлові затемнення та стільниковий малюнок у периферійних та нижніх ділянках легень [34].
Прийняття рішення стосовно призначення КТВРЗ пацієнту з задишкою та нормальною рентгенограмою органів грудної клітки є досить складним. Автори рекомендують візуалізаційні дослідження перерізів легень у випадку виявлення рестрикції чи низької ДЄМВ при визначенні фізіологічних функцій, а також при високому ступені підозри на хворобу легень як причину симптомів. У прийнятті цього рішення може допомогти експертна консультація, особливо коли причина задишки залишається нез’ясованою після проведення базових обстежень.
Якщо не вдається встановити діагноз за картиною КТВРЗ, необхідною є хірургічна біопсія з гістологічним підтвердженням картини ЗІП [16, 35]. Хоча бронхоскопія може бути цінною за умов підозри на інший діагноз (саркоїдоз, хронічний гіперчутливий пневмоніт), значення цього обстеження при діагностичному пошуку ІФЛ суперечливе. Плямиста природа змін при ЗІП може не виявитися у відносно малих зразках для біопсії, отриманих при бронхоскопії [36, 37].
У зв’язку з цим треба надавати перевагу хірургічному підходу до біопсії з використанням відкритої чи відеоасистованої торакоскопічної техніки. Остання є кращою як менш інвазивна процедура, яка не вимагає тривалого перебування у стаціонарі та супроводжується швидшим відновленням [38].
Бронхоскопічна кріобіопсія, яка на цей час досліджується, є потенційно цінним інструментом у діагностиці ІФЛ. Часто ні КТВРЗ, ні хірургічна біопсія легень не виявляє ЗІП, що робить встановлення точного діагнозу ІФЛ украй складним. Крім того, деякі пацієнти з неінформативною картиною при КТВРЗ не здатні перенести хірургічну біопсію легень у зв’язку з важкими порушеннями функції легень чи іншими коморбідними станами.
Коли хірургічна біопсія неможлива, для точного визначення діагнозу слід застосувати мультидисциплінарний підхід [30, 39]. У обговоренні всіх аспектів клінічного випадку мають брати участь пульмонологи, патологи, рентгенологи та інші спеціалісти з подальшим формуванням консенсусу щодо діагнозу та плану лікування.
Якщо у процесі дискусії робиться висновок про наявність ІФЛ, лікар може розпочинати терапію. Якщо ні, консультанти можуть рекомендувати подальші діагностичні обстеження чи пропонувати альтернативні діагнози та способи лікування. Ще одним завданням мультидисциплінарної команди є розгляд ризиків та переваг проведення хірургічної біопсії легень для окремих пацієнтів.
Лікування ІФЛ
Антифібротична терапія при ІФЛ (табл.) може включати пірфенідон чи нінтеданіб. Пірфенідон, молекулярна мішень якого точно невідома, був схвалений для цієї мети за результатами трьох досліджень [41, 42]. Узагальнені результати даних єкспериментів виявили сповільнення зменшення ФЖЄЛ та більш тривале виживання без прогресування хвороби [43]. Також при метааналізі клінічних випробувань пірфенідону було виявлено зменшення смертності, хоча жодне окреме дослідження такого результату не показало [44]. Головними побічними ефектами пірфенідону є порушення з боку шлунково-кишкової системи та поява фоточутливості з розвитком висипки.
Нінтеданіб є потрійним інгібітором тирозинкінази, мішенями впливу якого є фактор росту фібробластів, фактор росту ендотелію судин, рецептори тромбоцитарного фактора росту. Комбінований аналіз двох досліджень [45] показав, що нінтеданіб сповільнює зменшення ФЖЄЛ так само, як і пірфенідон. Головним небажаним ефектом, асоційованим з нінтеданібом, є діарея. Оскільки цей препарат пригнічує фактор росту ендотелію судин, існує також ризик гематологічних ускладнень, таких як кровотеча чи, навпаки, тромбози.
Через те що пірфенідон та нінтеданіб можуть підвищувати рівні амінотрансфераз, потрібно регулярно контролювати ці показники. Станом на сьогодні не існує дослідження, яке б порівнювало ефективність та переносимість пірфенідону та нінтеданібу. Отже, вибір засобу ґрунтується на побажаннях пацієнта після обговорення з ним потенційних ризиків, очікуваних переваг, профілів побічних ефектів. Треба також враховувати коморбідні стани та досвід лікаря. Пацієнти мають розуміти, що обидва ці засоби сповільнюють зниження ФЖЄЛ, однак не зменшують симптоми і не покращують функціональний стан.
Не варто рутинно застосовувати кортикостероїди (КС) у лікуванні ІФЛ. Хоча КС (у вигляді монотерапії чи у поєднанні з іншими імуносупресантами) широко застосовувалися для лікування ІФЛ у минулому, це використання не ґрунтувалося на результатах рандомізованих контрольованих досліджень [46]. Ретроспективні контрольовані дослідження не виявили, що КС зменшують рівні смертності у пацієнтів з ІФЛ, до того ж їхнє застосування супроводжується високими показниками фатальних наслідків [47, 48].
До того ж рандомізоване контрольоване дослідження, у якому КС поєднували з N-ацетилцистеїном та азатіоприном, було завчасно припинено у зв’язку зі збільшенням ризику смерті та госпіталізації [49]. У цілому наведені дані свідчать, що КС не забезпечують жодних переваг у лікуванні ІФЛ та є потенційно шкідливими, тому їхнє застосування при ІФЛ не рекомендовано. Об’єднані національні рекомендації взагалі не радять застосовувати імуносупресію в лікуванні ІФЛ [16].
Існують також інші керівні вказівки стосовно ведення пацієнтів з ІФЛ. Попередні дані свідчать, що мікроаспірація шлункового вмісту за умов гастроезофагального рефлюксу є фактором ризику ІФЛ. Тому існує слабка рекомендація агресивно лікувати у таких хворих гастроезофагальну рефлюксну хворобу [50].
Однак оскільки докази свідчать, що застосування інгібіторів протонової помпи може асоціюватися з побічними ефектами з боку нирок чи центральної нервової системи, слід підходити до цієї рекомендації з обережністю. Є припущення, що експерименти, які тривають на даний момент, забезпечать краще розуміння ролі кислотосупресії в лікуванні ІФЛ [51, 52].
Крім перелічених, рекомендації з лікування ІФЛ передбачають підтримуючу терапію (застосування кисню, легеневу реабілітацію, вакцинації). Обов’язковим є швидкий розгляд можливості пересадки легень. У наш час ІФЛ є найбільш поширеним показанням до трансплантації, і, враховуючи несприятливий прогноз при пізніх стадіях цього захворювання, пересадка легень надає суттєві переваги за умов правильного відбору пацієнтів [53, 54].
Загострення ІФЛ
У зв’язку зі своєю непередбачуваною природою ІФЛ може проявлятися у вигляді загострення без виявленої причини. Вільно трактовані критерії діагностики загострень ІФЛ включають діагноз ІФЛ (попередній чи вперше виявлений), погіршення чи розвиток задишки протягом останніх 30 днів, появу двобічних змін за типом матового скла чи консолідативних змін на тлі ЗІП при проведенні КТВРЗ [16].
Частота загострень ІФЛ є мінливою, у залежності від критеріїв ймовірність виникнення загострення впродовж 1-3 років становить 8,6-23,9% [56]. Загалом, загострення супроводжуються вкрай несприятливим прогнозом та медіаною очікуваної тривалості життя в межах 2,2 міс [57].
Отже, схваленої рекомендаціями терапії загострень ІФЛ не існує. Лікування є переважно підтримуючим і включає оксигенотерапію та механічну вентиляцію легень. Поточні настанови рекомендують КС (рекомендація слабкої сили), однак не вказують дозу, спосіб введення та тривалість лікування. Інші види лікування, передусім імуномодулятори, не мають достатньої доказової бази.
Список літератури знаходиться в редакції.
Tolle L. B., Southern B. D., Culver D. A., Horowitz J. C. Idiopathic pulmonary fibrosis: What primary care physicians need to know.
Cleveland Clinic Journal of Medicine. 2018 May; 85(5): 377-386.
Переклала з англ. Лариса Стрільчук
Медична газета «Здоров’я України 21 сторіччя» № 15-16 (436-437), серпень 2018 р.