


5 листопада, 2018
Сепсис: патогенез, імунодіагностика та імунотерапія (огляд та власні спостереження)
 В огляді звертається увага на порушення імунологічної функції при сепсисі, причини та механізми таких порушень та можливість імунокорекції як елементу терапії сепсису. Для імунокорекції важливо виявляти фазу реакції імунної системи організму на інфекцію, бо ті ж самі засоби можуть у різні фази імунної реакції бути як корисними, так і шкідливими. Для цього необхідно розробити та впровадити специфічні маркери. Також наводяться результати власних досліджень функції імунної системи людини при сепсисі.
В огляді звертається увага на порушення імунологічної функції при сепсисі, причини та механізми таких порушень та можливість імунокорекції як елементу терапії сепсису. Для імунокорекції важливо виявляти фазу реакції імунної системи організму на інфекцію, бо ті ж самі засоби можуть у різні фази імунної реакції бути як корисними, так і шкідливими. Для цього необхідно розробити та впровадити специфічні маркери. Також наводяться результати власних досліджень функції імунної системи людини при сепсисі.
Сепсис є складною клінічною проблемою, яка включає шкідливу або навіть руйнівну реакцію організму на інфекцію. Результати лікування сепсису залишаються незадовільними, летальність все ще тривожно висока, незважаючи на постійну розробку й впровадження нових методик лікування.
Стагнація в розробці нових антибіотиків (АБ), широке розповсюдження резистентних до АБ інфекційних патогенів та невдачі клінічних досліджень щодо корекції прозапальної фази сепсису є основними чинниками, які стимулюють дослідників у галузі сепсису розробляти та впроваджувати в клінічну практику застосування імуномодуляторів, які перш за все направлені на зменшення імуносупресії під час сепсису.
Метою цього огляду є зосередженість на імунопатогенезі й, особливо, на методах імуномодуляції сепсису.
Імунопатогенез сепсису
Історичною є думка, що сепсис складається з початкової гіперзапальної фази (синдром системної запальної відповіді – SIRS), з наступною протизапальною або імуносупресивною фазою (компенсаторний протизапальный синдром – CARS) [1, 2].
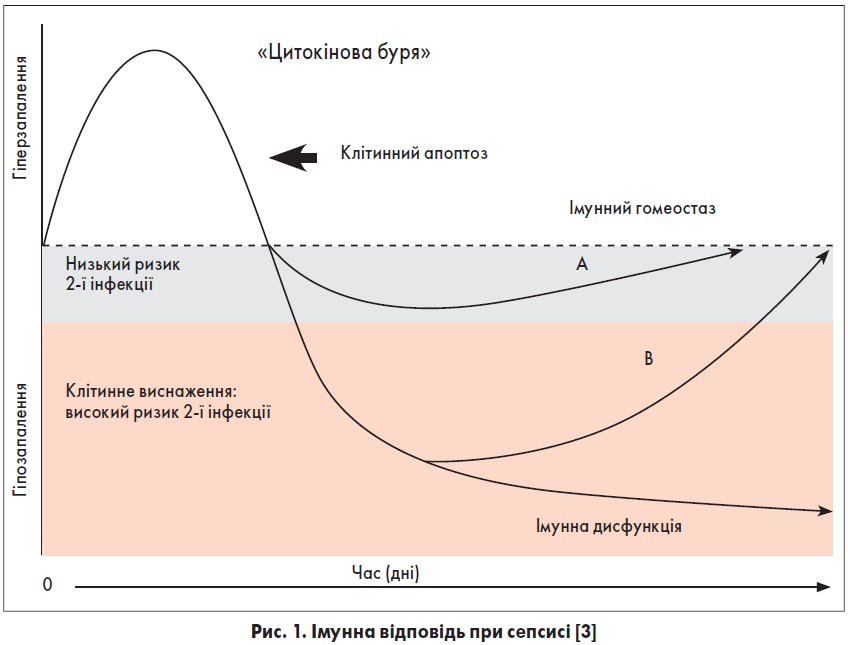 Імунна відповідь при сепсисі визначається багатьма чинниками, а саме: нутритивним статусом, загартуванням, супутньою патологією (цукровий діабет, захворювання серця, злоякісний процес), а також вірулентністю причинних патогенів септичного процесу. Хоча і про- й протизапальні механізми активуються одночасно вже на початку сепсису, протягом перших кількох діб у клінічній картині часто домінує гіперзапальна реакція.
Імунна відповідь при сепсисі визначається багатьма чинниками, а саме: нутритивним статусом, загартуванням, супутньою патологією (цукровий діабет, захворювання серця, злоякісний процес), а також вірулентністю причинних патогенів септичного процесу. Хоча і про- й протизапальні механізми активуються одночасно вже на початку сепсису, протягом перших кількох діб у клінічній картині часто домінує гіперзапальна реакція.
Гіперзапальну фазу назвали «цитокіновий шторм», який зумовлений підвищеними рівнями в плазмі прозапальних цитокінів TNF-α, IL‑1b та IL‑6. Але швидко відбувається виснаження і вроджених, і адаптивних імуноцитів внаслідок їх апоптозу, що знижує активність прозапальної реакції й призводить до імуносупресії (A). На цьому етапі для корекції імунного гомеостазу можливе застосування протизапальних засобів. Але паралельно або пізніше в пацієнтів може виникати неконтрольована протизапальна реакція з трансформацією в гіпозапальну фазу (B).
Якщо вона триває певний час, може виникнути клітинне виснаження; клітинний фенотип характеризується зниженою функцією Т-лімфоцитів, збільшенням активності експресії PD‑1 та зменшенням експресії ІL‑7R на Т-лімфоцитах. У цій фазі несприятливий результат лікування має місце в пацієнтів із неадекватною імунною відповіддю, зростає ризик вторинної інфекції, яку часто викликають умовно-патогенні збудники (рис. 1).
Наведена вище біфазна парадигма була заснована на багатьох дослідженнях, але сьогодні очевидно, що прозапальна та протизапальна фази можуть спостерігатися одночасно вже на ранній стадії сепсису.
Прозапальна реакція: цитокіновий «шторм»
Початкове імунне розпізнавання інфекційних патогенів пов’язане з молекулярними структурами (PAMPs), які виділяються з бактеріальних або грибкових організмів та раньового вмісту, що зв’язуються з рецепторами розпізнавання, які експресуються на активованих імунних клітинах [4, 5]. Активація патерн-розпізнавальних рецепторів призводить до продукції численних прозапальних молекул, включаючи TNF-α, ІL‑1β, ІL‑2, ІL‑6, ІL‑8, ІFN-γ, та протизапальних цитокінів, які викликають активацію та гальмування клітинних реакцій.
Ці реакції не обмежені посиленою фагоцитарною активністю і включають пошкодження судинного ендотелію з капілярною неспроможністю, синтез печінкою білків гострої фази, хемотаксис лейкоцитів до місця інфекції/запалення та активацію системи коагуляції [6, 7].
Вважається, що спочатку прозапальна реакція була основною причиною летальності у разі сепсису і, значною мірою, об’єктом для терапевтичних інтервенцій. Інтенсивність гіперзапальної початкової фази змінюється залежно від багатьох чинників, які включають початковий фізичний стан хворого, його супутню патологію, вірулентність патогенів, патогенне навантаження й генетичні фактори.
Результуючим ефектом запальної реакції, яка включає вроджений і адаптивний імунітет, є багаторазова циклічність цих фаз протягом сепсису [2]. Така біфазна уява, можливо, є спрощеною інтерпретацією складної хвороби, але все ж забезпечує раціональне пояснення змінення функцій імунної системи під час сепсису.
Адаптивний імунітет
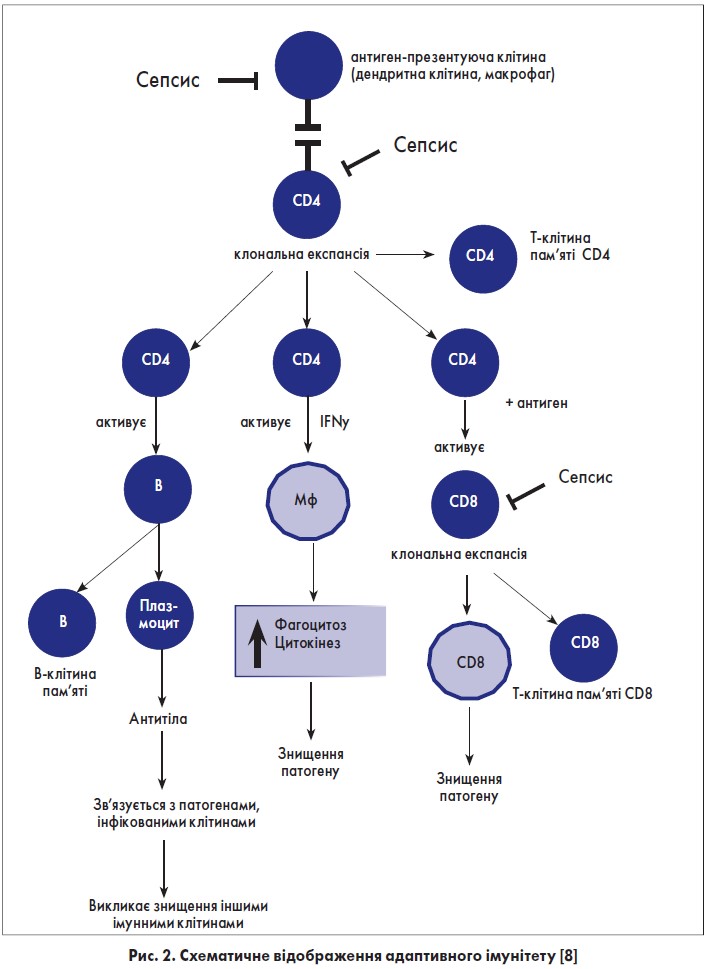 На рисунку 2 наведено спрощений набір різних клітин адаптивної імунної системи. Т-лімфоцити відіграють важливу роль в елімінації інфекційних патогенів [8].
На рисунку 2 наведено спрощений набір різних клітин адаптивної імунної системи. Т-лімфоцити відіграють важливу роль в елімінації інфекційних патогенів [8].
СD4+ Т-клітини хелперів-помічників, які активуються внаслідок презентації антигену антигенпрезентуючими клітинами. Активовані СD4+ Т-клітини розповсюджуються внаслідок клональної експанзії з утворенням клітин пам’яті СD4+ Т та ефекторних СD4+ Т-клітин. Ефекторні СD4+ Т-клітини сприяють активації В-клітин, СD8+ Т-клітин та навіть макрофагів, які беруть участь в елімінації патогенів. Сепсис порушує функцію як антигенпрезентуючих клітин, так і клітин системи адаптивного імунітету (СD4+ Т-лімфоцити, В-лімфоцити, СD8+ Т-лімфоцити та Мf – макрофаги).
Вроджені імуноцити – це дендритні клітини, макрофаги і моноцити, які активують наївні Т-лімфоцити у відповідь на антигенну стимуляцію, піддаються їх клональному розширенню. Вони пізніше продукують цитокіни, специфічні для антигена [9]. За умови позитивного перебігу інфекції гине більшість ефекторних Т-лімфоцитів (фаза скорочення), і ті Т-лімфоцити, які вижили, перетворюються на Т-лімфоцити пам’яті, важливі для ініціації реакції-відповіді в майбутньому на певні антигени після попереднього контакту з подібними антигенами.
У зумовленій антитілами відповіді, яку підтримують В-лімфоцити, беруть участь CD4+ Т-лімфоцити [10]. Антитіла (імуноглобуліни), які були продуковані В-лімфоцитами, є специфічними для певного антигена. Взаємодія імуноглобуліна та антигена призводить до багатьох ефектів, включаючи: інактивацію вірусів чи мікробних токсинів, блокування їх взаємодії з клітинами господаря, маркування інфекційних агентів для деструкції їх фагоцитами. Тому і Т-, і В-лімфоцити відіграють критичну роль у захисті хазяїна проти небезпечних для життя інфекцій. Послаблення таких критичних захисних механізмів веде до нездатності знешкодити основні інфекційні вогнища, що призводить до підвищеної чутливості до вторинних інфекцій під час сепсису [3].
Вважали, що гіперзапальна реакція, яка характеризується надмірною продукцією прозапальних медіаторів, таких як TNF-α та IL‑1, була ознакою сепсису і найбільш життєздатною терапевтичною метою численних преклінічних і клінічних досліджень [1]. Клінічні дослідження включали застосування антагоністів TNF та IL‑1b, блокаторів Толл-подібних рецепторів (TLR), інгібіторів тромбоцит-активуючого фактору (Xigris 12-14), антикоагулянтів, антагоністів ендотоксину, гемофільтрацію з метою видалити розчинні ендотоксини та цитокіни.
Всі вони в кінцевому підсумку не виявили ефективності, а в деяких випадках погіршували результат лікування сепсису [3, 11, 12]. Клінічні дослідження, в яких використовували блокатори прозапальних медіаторів, таких як антиендотоксинові антитіла до ліпополісахариду (ЛПС), прозапальні цитокіни (TNF-α, IL‑1β) та антагоністи TLR, показали невтішні результати [13, 14].
Отже, можливості поточного лікування сепсису обмежені. Вони головним чином включають антибіотикотерапію, відновлення об’єму рідини й підтримку органних систем. Така стратегія зменшила ранні смертельні випадки серед септичних пацієнтів та покращила загальну виживаність [15]. Але, незважаючи на ці успіхи, летальність у разі септичного шоку все ще перевищує 30% [16].
Гіпозапальна реакція: імунний «параліч»
Після запеклих дебатів було досягнуто угоди – сепсис може розвиватися в дві фази: перша – гіперзапалення (цитокіновий «шторм») і друга – гіпозапалення (імунний «параліч»), як показано на рисунку 1 [2, 17]. Багато доказових досліджень та краще розуміння імунологічних альтерацій під час сепсису протягом останнього десятиріччя довели роль імуносупресії в патогенезі сепсису [18, 19].
Останнім часом імуносупресія, яка виникає на тлі сепсису, є темою інтенсивних досліджень численних авторів у всьому світі. Дійсно, у різних дослідженнях доведено, що пацієнти з початковою запальною фазою сепсису дуже чутливі до шпитальних інфекцій, причиною яких є умовно-патогенні організми, і серед хворих, які початково вижили після сепсису, спостерігають пізні смертельні випадки [20, 21].
Хоча сучасні стратегії лікування короткостроково покращували стан септичних пацієнтів, всі вони однаково супроводжувалися більш тривалою зміною на імуносупресивний фенотип, який збільшує кількість відтермінованих смертей. Фактично, багато летальних випадків стаються після 3 діб від початку сепсису, а значна частина – протягом кількох тижнів від початку сепсису [22]. На думку деяких вчених, імунотерапевтичні стратегії, направлені на стимуляцію імунної системи, мають значний потенціал, який здатний змінити викликану сепсисом імуносупресію та покращити стан пацієнта.
Пацієнти відповідають на сепсис гетерогенним чином: продукцією прозапальних і протизапальних цитокінів у ранньому періоді після початку сепсису, у декотрих – із наступною зниженою секрецією цитокінів або однією тільки протизапальною реакцією [2]. Стало очевидно, що в багатьох септичних хворих, які пережили гіперзапальну початкову фазу – «цитокіновий шторм», розвивається відтермінована і потенційно тривала компенсаторна протизапальна відповідь, так званий компенсаторний протизапальний синдром (CARS) [2, 22].
Хоча деякі хворі все ще помирають під час початкової фази сепсису, нові протоколи лікування сприяють рятуванню більшості пацієнтів у цій фазі, після якої розвивається стан імуносупресії [15]. Ця імунна дисфункція стає причиною підвищеної чутливості організму до вторинних бактеріальних інфекцій, наприклад вентилятор-асоційованої пневмонії (VAP), інфекцій, які викликані, взагалі, авірулентними або умовно-патогенними організмами. При цьому зростає ризик розвитку CARS [23].
Численні дослідження показали, що в результаті багатьох імунних порушень гіпозапалення призводить до дисфункції адаптивної імунної відповіді [24-28].
Адаптивна імунна система складається зі спеціалізованих антиген-специфічних клітин, важливих для генерування клітин пам’яті при повторних реакціях на антигени. CD4+ і CD8+ є головними субпопуляціями Т-лімфоцитів. CD4+ Т-лімфоцити відомі як Т-хелпери, і вони відіграють значну роль в оркеструванні численних реакцій вродженої та адаптивної імунних систем [10]. CD8+ Т-лімфоцити-кілери, також відомі як цитотоксичні Т-лімфоцити, важливі для знищення пухлинних або інфікованих клітин [29]. Специфічні для конкретного антигену Т-лімфоцити необхідні для стимуляції продукції антитіл проти внутрішньоклітинних (Th1) або позаклітинних (Th2) патогенів за участю розчинних медіаторів-цитокінів.
Продукцію всіх антитіл, які належать до гуморального імунітету, забезпечують В-лімфоцити за допомоги Т-лімфоцитів. Специфічне для антигена антитіло може нейтралізовувати токсини, фіксувати комплемент та визначати поверхню для фагоцитарного поглинання інфекційних патогенів моноцитами/макрофагами. Антигенпрезентуючі клітини, які включають моноцити/макрофаги та дендритні клітини, аналізують середовище і презентують антигени Т-лімфоцитам для ініціації імунної відповіді або індукції толерантності адаптивного імунітету.
Збільшується кількість даних, які підтверджують гіпотезу, що імуносупресія відіграє важливу роль у летальності, зумовленій сепсисом [3]. Імуносупресія під час сепсису пояснюється тим, що септичні хворі дуже чутливі до вторинних інфекцій: Pseudomonas, Candida, Acinetobacter, Enterococcus, а також високою частотою реактивації персистуючих вірусів, таких як цитомегаловірус, вірус простого герпесу, вірус Епштейна – Барр [30-32].
Цікаві клінічні спостереження, а також імунологічні та молекулярно-генетичні дослідження були проведені львівською школою хірургів у 2013-2016 рр. за участю 129 хворих з абдомінальним сепсисом [33, 34]. Вони показали розвиток швидкої імуносупресії в септичних хворих, особливо з тяжким абдомінальним сепсисом, на основі аналізу кількості активованих Т-лімфоцитів (CD3+HLA-DR+), регуляторних лімфоцитів (CD4+CD25brig+CD127neg) та імунорегуляторних молекул мікро-РНК 146а та 155 вродженого й адаптивного імунітету.
Так, у таблиці 1 представлено порівняльну характеристику показників активованих Т-лімфоцитів та їх регуляторної субпопуляції у хворих на гострі хірургічні захворювання органів черевної порожнини з абдомінальним сепсисом (ГХЗОЧП + АС) та тяжким АС.
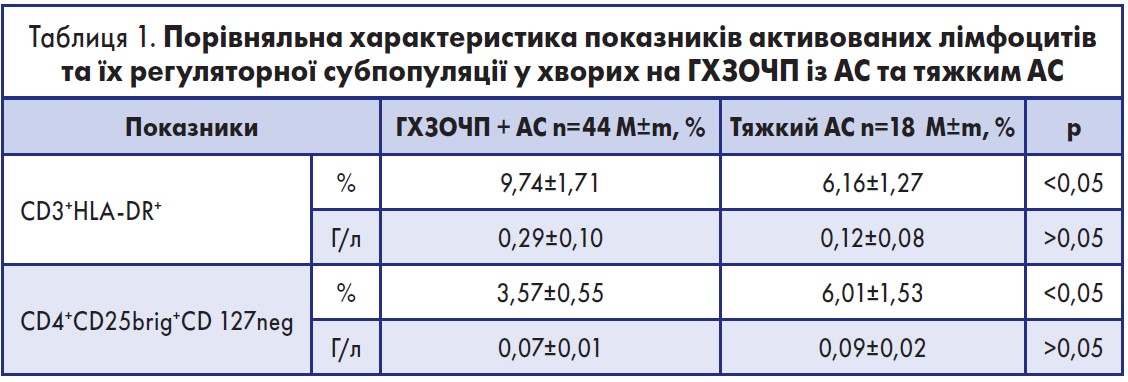
Отже, у досліджуваних хворих із тяжким АС виявлено вірогідно меншу відносну кількість активованих лімфоцитів порівняно з хворими на ГХЗОЧП + тяжкий АС (р<0,05) та збільшення в них відносної кількості регуляторних лімфоцитів (р<0,05). Отримані дані свідчать про наростання імуносупресії у хворих із тяжким АС, що відповідає доказовим дослідженням [19].
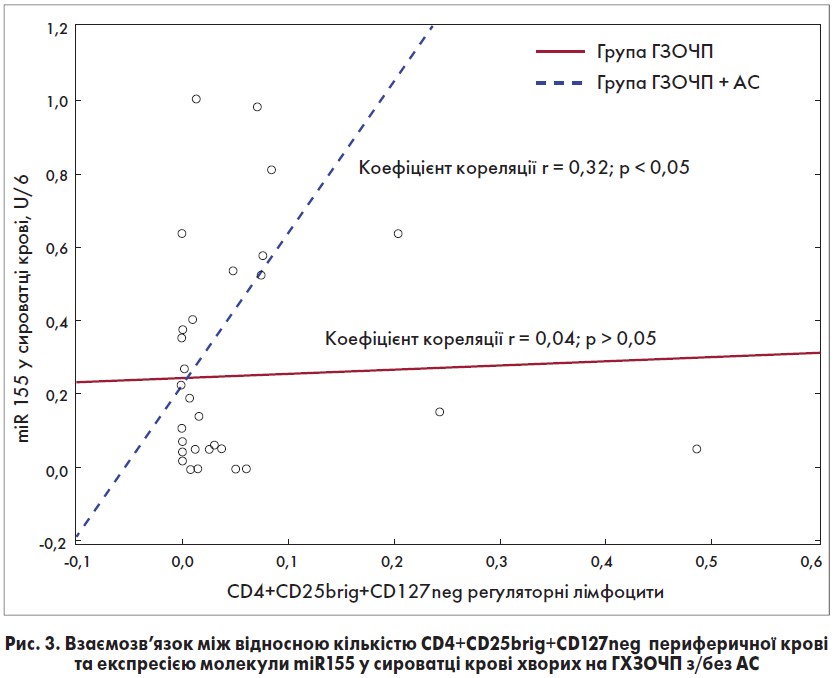 Крім того, було встановлено взаємозв’язок між рівнем експресії miR-155 у сироватці крові (це прозапальний епігенетичний біомаркер, що впливає на активність адаптивної клітинної відповіді імунної системи) та кількістю регуляторних Т-лімфоцитів у периферичній крові, який показав пряму кореляцію між наростанням стимулюючого фактора імунної лімфоцитарної активності та клітинної імуносупресії (r=0,32) у всіх хворих з АС, та відсутність взаємозв’язку у хворих без сепсису, як показано на рисунку 3. Це свідчить про ранній розвиток імуносупресивної фази сепсису.
Крім того, було встановлено взаємозв’язок між рівнем експресії miR-155 у сироватці крові (це прозапальний епігенетичний біомаркер, що впливає на активність адаптивної клітинної відповіді імунної системи) та кількістю регуляторних Т-лімфоцитів у периферичній крові, який показав пряму кореляцію між наростанням стимулюючого фактора імунної лімфоцитарної активності та клітинної імуносупресії (r=0,32) у всіх хворих з АС, та відсутність взаємозв’язку у хворих без сепсису, як показано на рисунку 3. Це свідчить про ранній розвиток імуносупресивної фази сепсису.
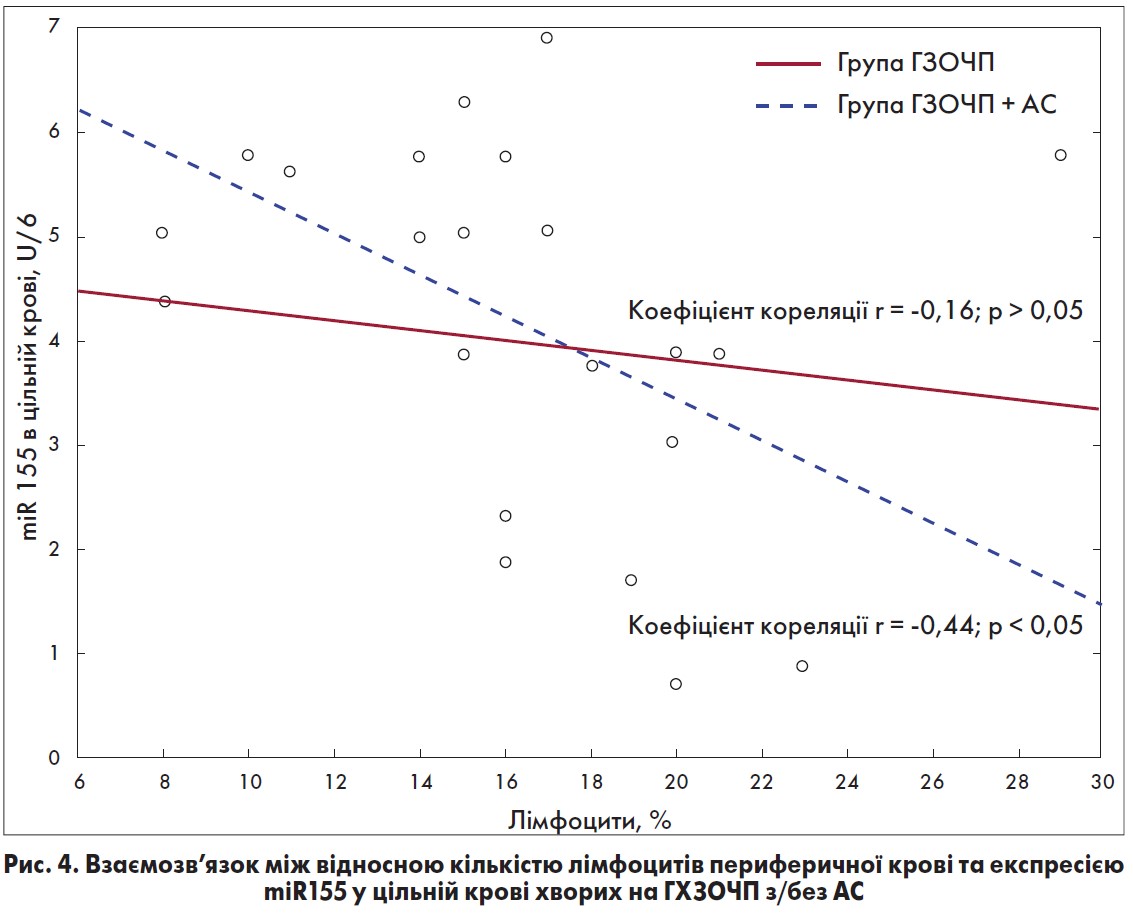
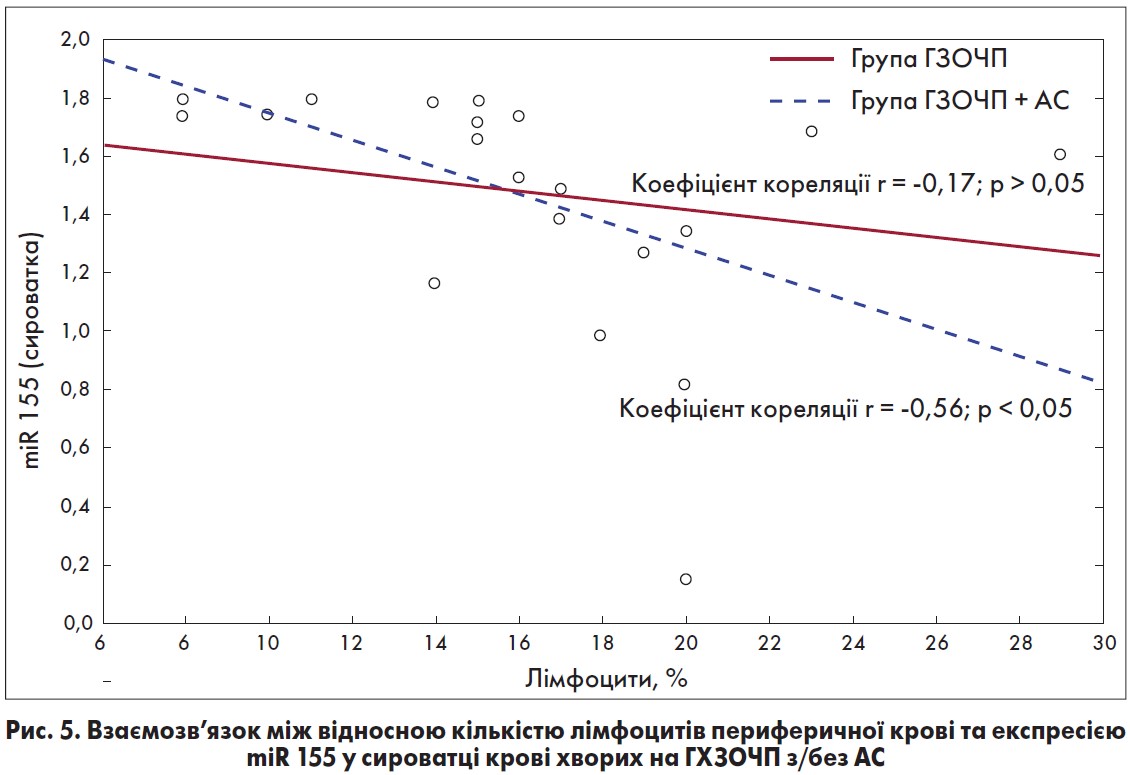
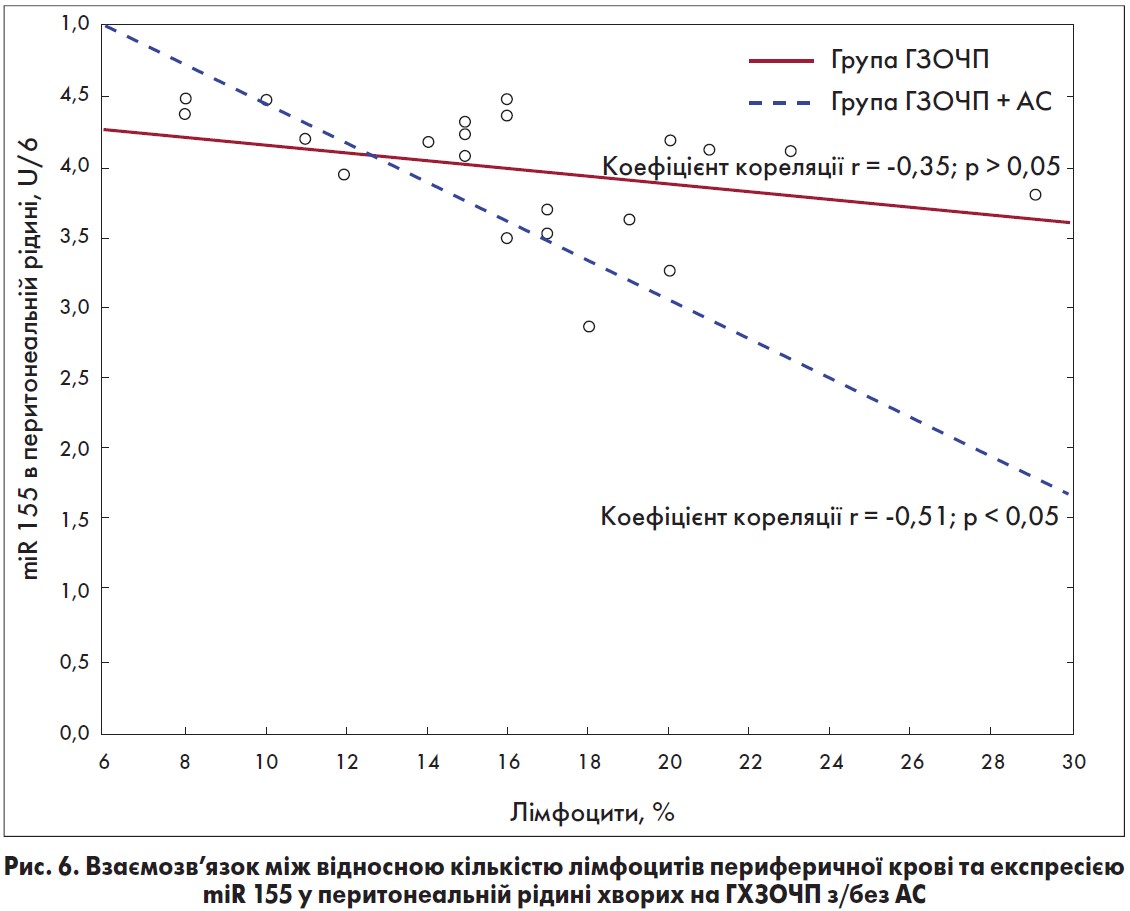
На рисунках 4-6 показаний взаємозв’язок між відносною кількістю лімфоцитів периферичної крові та експресією miR-155 у цільній крові, сироватці крові та перитонеальній рідині в досліджуваних хворих. Як бачимо, у хворих на ГХЗОЧП, що не були ускладнені АС, були виявлені незначні обернені зв’язки між кількістю лімфоцитів периферичної крові та експресією miR-155.
Водночас у септичних пацієнтів були встановлені виражені обернені взаємозв’язки між відносною кількістю лімфоцитів периферичної крові та експресією miR-155 у біологічних середовищах: r= -0,44 – у цільній крові; r= -0,56 (р<0,05) – у сироватці крові та r= -0,51 – у перитонеальній рідині [35]. Отримані результати дають підстави припустити, що процеси імуностимуляції та імуносупресії відбуваються паралельно.
У таблиці 2 наведено порівняльну характеристику показників активованих лімфоцитів та їх регуляторної субпопуляції у хворих на гострі хірургічні захворювання черевної порожнини, що ускладнені абдомінальним сепсисом, які вижили, та хворих, що померли.
Як видно з таблиці 2, відносна кількість активованих лімфоцитів у пацієнтів, що померли в результаті септичних ускладнень, була значно нижчою порівняно із септичними пацієнтами, що вижили (р<0,05).
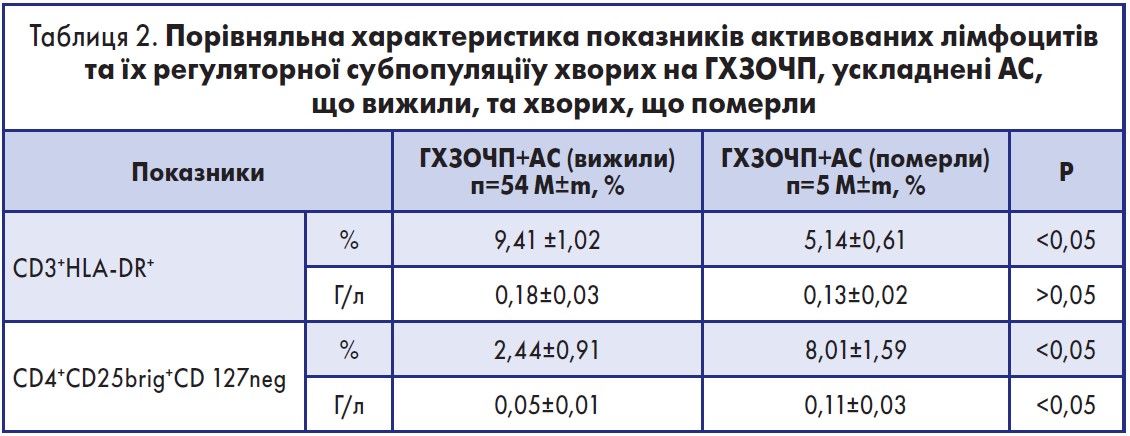
У померлих хворих відзначено значуще підвищення абсолютної та відносної кількості регуляторних лімфоцитів порівняно з тими пацієнтами, що вижили (р<0,05). Поясненням цьому може бути значна депресія клітиннозалежної та гуморальної імунної відповіді з формуванням набутого імунодефіцитного порушення, пов’язаного із сепсисом [36].
Аналогічно, за допомогою методу логістичної регресії було виокремлено також 7 чинників, які при поєднаній дії мають вірогідний вплив на розвиток тяжкого сепсису. Значення коефіцієнтів регресії для вказаних чинників наведено в таблиці 3.
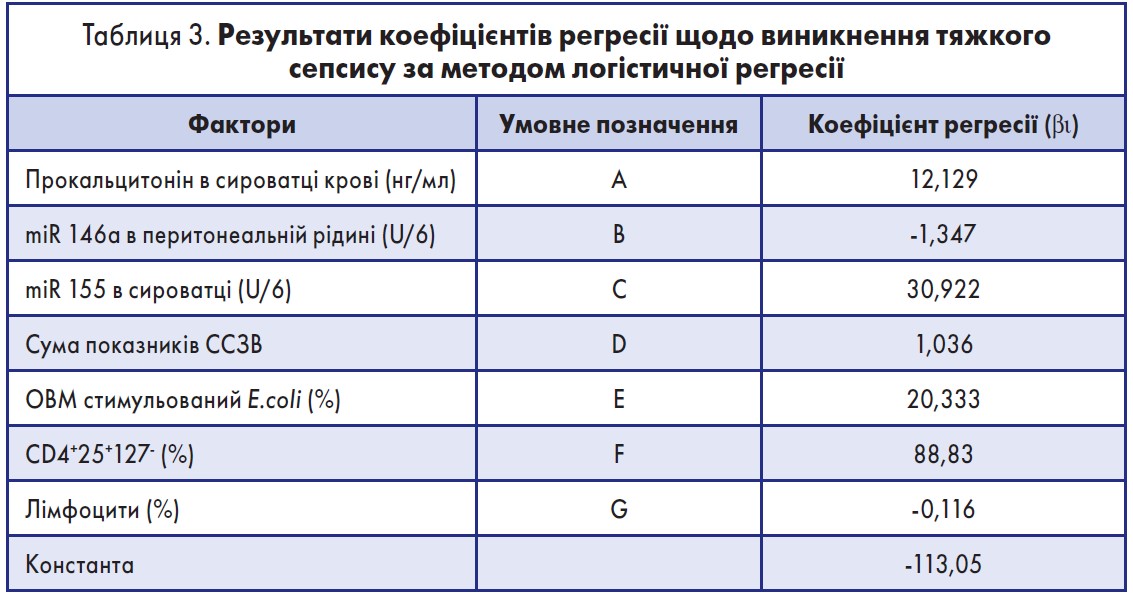
Підставивши у формулу результати, отримані за методом логістичної регресії, ми отримали значення Z для визначення вірогідності розвитку тяжкого сепсису:
Z = 12,129A – 1,347 B + 30,922 C + 1,036 D + 20,333 E + 88,830 F – 0,116G – 13,050
(6.2), де:
A – прокальцитонін у сироватці крові;
B – miR-146а у перитонеальній рідині – епігенетичний фактор активації природженого імунітету;
C – miR-155 у сироватці крові – епігенетичний фактор активації адаптивного імунітету;
D – сума показників синдрому системної запальної відповіді;
E – ОВМ стимульований E. coli – оксидантний «вибух» стимульованих E. coli моноцитів;
F – CD4+25+127- – регуляторні Т-лімфоцити;
G – лімфоцити.
Відтак превентивними чинниками щодо розвитку тяжкого сепсису є концентрація miR-146а в перитонеальній рідині та кількість лімфоцитів у крові. Всі інші ознаки – провокуючі. Також була проведена ретроспективна перевірка отриманої моделі на основі результатів дослідження. Отримані результати засвідчують, що точність регресійного рівняння щодо прогнозування тяжкого сепсису становить 98,8% [37]. Отже, проведене дослідження, що поряд із запальною фазою, яку характеризує синдром системної запальної відповіді (SIRS), розвивається імуносупресивна фаза сепсису – компенсаторний протизапальний синдром (CARS).
Імуносупресія при сепсисі
Головними механізмами, які призводять до імуносупресії при сепсисі, є погіршення функції адаптивної імунної системи, що включає:
- прискорений апоптоз, який призводить до виснаження клітин;
- погіршення реактивності внаслідок посиленої регуляції інгібуючих рецепторів або низькорівневої регуляції есенціальних костимулюючих рецепторів на поверхні клітин;
- зниження реакції цитокінів [31, 38].
Сепсис-індукований апоптоз клітин адаптивного імунітету
 Апоптоз, відомий також як «програмована смерть» клітин, є одним із шляхів, за допомогою яких імунна система підтримує гомеостаз, ліквідуючи активовані та відпрацьовані клітини. Цей термін почав використовуватися в історичній роботі J. Kerr і співавт. (1972), в якій було описано різні форми некрозу клітин [39].
Апоптоз, відомий також як «програмована смерть» клітин, є одним із шляхів, за допомогою яких імунна система підтримує гомеостаз, ліквідуючи активовані та відпрацьовані клітини. Цей термін почав використовуватися в історичній роботі J. Kerr і співавт. (1972), в якій було описано різні форми некрозу клітин [39].
Апоптоз – фізіологічний процес, що протікає під час нормального росту та розвитку клітин, але він може набути шкідливих наслідків для виживання організму за багатьох патологічних станах, у тому числі при сепсису [40, 41].
Центральними регуляторами апоптозу є каспази – цистеїнові протеази. Вони сприяють деградації клітинних білків та ядерного фактору NFкB – транскрипційного фактору, який активує транскрипцію обох генів – і проапоптичного, і антиапоптичного. Гіперзапальна реакція при сепсисі для продукції прозапальных цитокінів потребує активації NFкB та каспаз, при цьому і NFкB, і каспази одночасно викликають апоптоз адаптивних імунних клітин [42, 43]. Апоптоз при сепсисі, як показали вчені, виникає паралельно з прозапальною реакцією. У моделях сепсису на мишах, так само як у хворих із тяжким сепсисом, виникає значна деплеція T-, B- і дендритних клітин [44-47].
У межах перших 24 год після початку сепсису у хворих виникає лімфопенія, яка розвивається внаслідок рекрутування лімфоцитів із кровообігу у вогнища запалення/інфекцій та апоптичної деплеції CD4- та CD8-лімфоцитів у крові [44]. Посмертний аналіз селезінок та лімфатичних вузлів у померлих від сепсису підтвердив значущу втрату CD4-- та CD8--лiмфоцитiв [45]. Клітини пам’яті та CD8-лiмфоцити дуже чутливі до апоптозу за умов системного запалення, таких як септичний шок. Вони швидко виснажуються за цих умов [48].
Результати досліджень демонструють, що виснаження адаптивних імуноцитів – головний патологічний компонент сепсису з потенційними несприятливими ефектами для імунної системи організму.
Апоптоз – дуже виражений патологічний процес при сепсисі. Так, у дослідженні S. Wang та співавт. ін’єкція грамнегативних бактерій мишам призводила до апоптозу CD4+- та CD8+-клітин у тимусі [49]. У 1990-х роках в інших дослідженнях було визначено потужний апоптоз лімфоцитів у різних органах, включаючи селезінку, тимус, товсту та тонку кишку, легені та скелетні м’язи при сепсисі, що був спричинений перев’язкою та пункцією сліпої кишки (CLP) у мишей [50].
 В іншому дослідженні визначили апоптоз B22+-клітин у кістковому мозку та незрілих Т-лімфоцитів у тимусі [51]. Після цього численні преклінічні дослідження представили точні підтвердження того, що сепсис призводить до значної, викликаної апоптозом, втрати CD4+- та CD8+-лімфоцитів [52-55].
В іншому дослідженні визначили апоптоз B22+-клітин у кістковому мозку та незрілих Т-лімфоцитів у тимусі [51]. Після цього численні преклінічні дослідження представили точні підтвердження того, що сепсис призводить до значної, викликаної апоптозом, втрати CD4+- та CD8+-лімфоцитів [52-55].
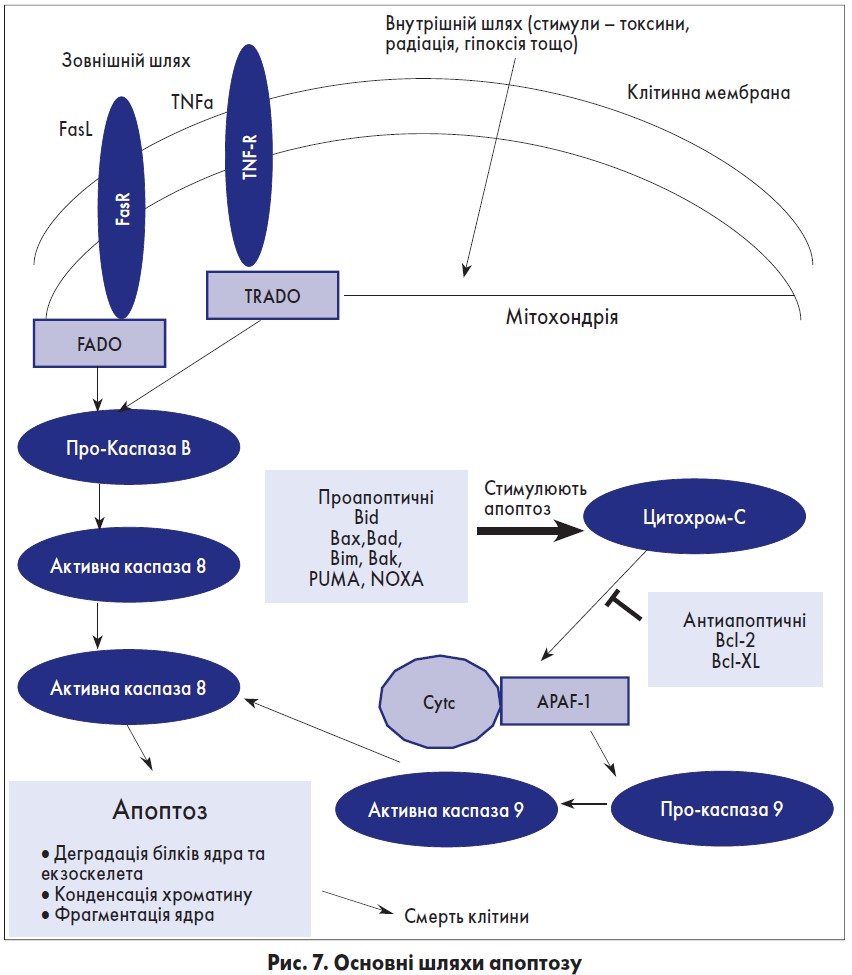
Значне виснаження Т-лімфоцитів у селезінці та лімфатичних вузлах також знайшли при системній інфекції Pseudomonas під час масивних опіків [56]. Хоча деплеція адаптивних імуноцитів визнана важливим компонентом патогенезу сепсису, механізми, відповідальні за це, не повністю зрозумілі [57, 58]. На рисунку 7 наведені основні шляхи апоптозу.
Зовнішній шлях апоптозу реалізується через взаємодію головних «смертельних» лігандів, таких як Fas (FasL або CD95L) та TNF-α з відповідими рецепторами «смерті», такими як рецептор Fas (FasR або CD95R) і рецептор TNF (TNFR) відповідно, на поверхні клітин. Взаємодія Fas/FasR призводить до закріплення білків адаптерів FADD та TRADD, із наступною дією FADD із рецептором взаємодіючого білка (RIP).
Внутрішній шлях апоптозу включає порушення цілісності мембрани мітохондрій під впливом різноманітних стимулів, таких як бактеріальні токсини, радіація та ін. Токсичні стимули порушують цілісність мембран мітохондрій, що призводить до відкриття мітохондріальних пор перехідної проникності (MPTР), втрати мембранного потенціалу та виходу мітохондріального цитохрому в клітинну цитоплазму, що в кінцевому підсумку призводить до активації каспази‑9, яка розщеплює каспазу‑3, призводячи до реалізації апоптозу та некрозу клітин [59-61]
Регуляторні Т-лімфоцити здатні пригнічувати проліферацію Т-лімфоцитів, секрецію цитокінів і, знову ж таки, сприяти апоптозу Т-лімфоцитів [62]. У септичних пацієнтів відсоток таких клітин збільшується в результаті їх стійкості до апоптозу та зменшення кількості активованих T-клітин, що є ще одним потенційним механізмом імунної дисфункції.
Експресія цих інгібуючих рецепторів/лігандів на Т-лімфоцитах, антигенпрезентуючих клітинах та тканинах є середовищем, яке сприяє виснаженню Т-лімфоцитів і анергії імунітету в септичних пацієнтів [63, 64]. Однак кінцевим виконавцем усіх апоптичних шляхів є активація каспази‑3 – вона викликає фрагментацію ДНК та розпад цитоскелетних, а також ядерних білків, що призводить до некрозу клітин [65, 66].
Одним із основних білків, що контролюють мітохондріальний шлях апоптозу, є родина Bcl‑2 білків, які керують вивільненням цитохрому шляхом регуляції мітохондріальної мембранної проникності. Декотрі з антиапоптичних білків належать до родини Bcl‑2, до якої включені наступні молекули: Bcl‑2, Bcl-x, Bcl-XL, Bcl-XS, Bcl-w, BAG. До проапоптичних білків належать: Bcl‑10, Bax-, Bid-, Bim-, Bik-, Bad- і Blk-молекули.
Ці білки за участю різних механізмів викликають апоптоз. Так, нефосфорильована форма Bad може переміститися в мітохондрії та викликати вивільнення цитохрому, також може зв’язуватися з Bcl-Xl та Bcl‑2 білками, таким чином інгібувати їхні захисні ефекти [67, 68]. Хоча механізм цього процесу не зовсім зрозумілий, Bcl‑2 та Bcl-XL інгібують апоптоз, регулюючи активацію каспаз [69].
Мітохондріальне пошкодження зустрічається при Fas-залежному зовнішньому шляху апоптозу через розпад Bid, який активується каспазою‑8. Він може переміститися в мітохондрії, де зв’язується з Вах/Вак, що теж призводить до апоптозу [70]. Цей механізм – один із відомих ланцюгів інтеракції між двома класичними шляхами апоптозу [71]. Для обох (і внутрішнього, і зовнішнього) шляхів апоптозу активація каспази‑3 оркеструє заключний етап апоптозу з некрозом клітин.
Втрата лімфоцитів визначається балансом про- та протизапальних механізмів. У посмертному дослідженні сепсису було зареєстровано зменшення експресії СD28 на Т-лімфоцитах у септичних пацієнтів порівняно з пацієнтами контрольної групи [45]. Ко-стимулююча молекула СD28, якщо вона зв’язується з молекулами В7 на антигенпрезентуючих клітинах, потребує продукції ІL‑2 та експресії Всl-ХL. Ці два фактори свідчать про антиапоптичний ефект та виживання Т-клітин [72].
Антигенпрезентуючі клітини, включаючи дендритні клітини та макрофаги, виділені або з селезінки, або з легень септичних хворих, мали низьку експресію молекул В7(В7-1/СD80 та В7-2/СD86), що потенційно обмежувало здатність Т-лімфоцитів до ко-стимуляції. Без необхідної поєднаної стимуляції через СD28/В7, коли рецептори Т-лімфоцитів залучають антиген, Т-лімфоцити піддаються процесу, який називають «відсутність функціональної активності» (анергія).
Недавні дослідження також показали, що Т-лімфоцити позитивно регулюють поверхневі рецептори, котрі інгібують функції Т-лімфоцитів. Було ідентифіковано збільшення експресії програмованого рецептора «смерті» 1(РD‑1), СТLА‑4, В-атенюатора (ВТLА) на Т-лімфоцитах, які були виділені посмертно із селезінки та легень, а також із крові через 7 днів після постановки діагнозу «сепсис».
Крім того, було визначено збільшення експресії програмованих лігандів смерті -1 та -2 (РD-L1 та РD-L2) на антигенпрезентуючих клітинах, виділених із крові через 7 днів після постановки діагнозу «сепсис» [44, 45]. Експресія РD-L на антигенпрезентуючих та ендотеліальних клітинах була пов’язана з індукцією імунної толерантності [73, 74].
Збільшення експресії молекул PD‑1, СTLА‑4 або ВTLА (усі – члени суперродини CD28-рецепторів) викликає апоптоз Т-лімфоцитів або клітинну толерантність, що сприяє нормалізації імунного гомеостазу. PD-L та СTLА‑4 молекули, так само як і члени родини ліганду TNFR та Fas-ліганд – CD95L), експресуються на спеціалізованому типі CD4+-лімфоцитів, які вже самі експресують транскрипційний фактор FoxP3 та поверхневу CD25-молекулу (IL‑2Rα). Ці клітини було названо регулюючими Т-лімфоцитами (Тreg) [75].
Було доведено, що інгібування апоптозу з використанням специфічних інгібіторів каспаз, зокрема каспази‑3 у мишей, або збільшена експресія антиапоптичного білка Вс1-2 значуще зменшували апоптоз Т-лімфоцитів і покращували виживання тварин у моделі CLР сепсису [76, 77].
Ще одне важливе спостереження полягало в тому, що у Т- і В-лімфоцитодефіцитних мишей (Rаg-/-knockout) спостерігали підвищену летальність при сепсисі порівняно з диким типом мишей [76]. Інгібітори каспаз не надавали переваг для виживання при сепсисі, це було підставою вважати, що адаптивна імунна відповідь важлива для виживання при сепсисі. Подібні результати преклінічного дослідження також було підтверджено в клінічних дослідженнях септичних пацієнтів.
Виснаження Т-клітин
Виснаження Т-лімфоцитів – це патологічний стан, який часто характеризується втратою ефекторних функцій, збільшеною регуляцією численних інгібуючих рецепторів на поверхні клітин, низькорівневою регуляцією ко-стимулюючих рецепторів, зміною експресії ключових транскрипційних факторів та метаболічними розладами.
Таке виснаження спостерігається у хворих на ВІЛ-інфекцію, вірус гепатиту С і рак [78]. Ці процеси було описано в мишей із хронічними вірусними інфекціями [79]. У виснажених Т-лімфоцитів знижена функціональна здатність зі зменшеною продукцією цитокінів (γ-інтерферону, IL‑2, гранзиму-В, TNF-α), у них порушена проліферація, вони більш схильні до апоптозу.
Головні інгібуючі рецептори на поверхні Т-лімфоцитів включають: PD‑1, атенюатори Т- і В-лімфоцитів (ВTLА або CD272), цитотоксичний Т-лімфоцит-асоційований білок‑4 (СTLА‑4 або CD152), білок мембрани Т-лімфоцитів 3 (ТІМ‑3), та лімфоцит-активований ген‑3 (LAG‑3). Ліганди для цих гальмуючих рецепторів зазвичай присутні на антигенпрезентуючих клітинах, таких як дендритні клітини, макрофаги й моноцити, а на цільових клітинах – безпосередньо. PD‑1 взаємодіють з програмованими лігандами смерті ‑1 і -2 (PD-L1, PD-L2), BTLA взаємодіють з HVEM (медіатор розвитку вірусу герпесу), TIM‑3 із CEACAM‑1 (карцино-ембріональна, пов’язана з антигеном адгезивна молекула -1) або з галактин‑9, LAG‑3 взаємодіє з молекулою антигена класу основного комплексу гістосумісності (MHC), молекула CTLA‑4 взаємодіє з CD80/CD86 на антигенпрезентуючих клітинах [60].
Ко-стимулюючі сигнальні молекули CD28 на поверхні клітин знаходяться поряд із рецептором Т-лімфоцитів (TCR), і вони потрібні для оптимальної функції Т-лімфоцитів, включаючи проліферацію, виживання клітин та продукцію IFN-γ, IL‑2, які є важливими для боротьби з інфекціями [80].
У доповнення до апоптозу виснаження Т-лімфоцитів, яке веде до зниження реактивності, відіграє ключову роль у патофізіології імуносупресії, спричиненої сепсисом [2, 81].
Виснаження Т-лімфоцитів часто розвивається при персистуючій присутності антигена та/або запаленні [82]. Високе антигенне навантаження та запалення, викликане інфекцією, є характерними особливостями сепсису [83]. Саме тому у Т-лімфоцитів є пролонгована здатність реагувати на персистуючі стимули, котрі можуть призвести до збільшеної експресії на поверхні клітин інгібуючих рецепторів.
Це ще раз підкреслює необхідність адекватної санації інфекційного вогнища.
PD-L1 є одним із головних лігандів, які взаємодіють з інгібуючими рецепторами під час сепсису. PD‑1 зазвичай позитивно регулюються на поверхні CD4+ та CD8+ Т-лімфоцитів у відповідь на стимуляцію, щоб попередити надмірну активацію Т-лімфоцитів [82, 84].
Низькорівневі сигнали PD‑1, які призводять до дисфункції Т-лімфоцитів, включають внутрішньоклітинний імунний рецептор тирозин-залежний тригер (ITSM) та імунний рецептор, який оснований на тирозин-пригнічуючих властивостях (ITIM) і рекрутує тирозин-білкові фосфатази SHP1 та/або SHP2 (відповідність SRC2 області, яка містить фосфатази).
Численні преклінічні дослідження показали тривале збільшення експресії PD‑1 та PD-L1 під час сепсису. У моделях на тваринах сепсис викликав збільшену експресію PD‑1 на Т-лімфоцитах та PD-L1 експресію на моноцитах, яка пов’язана зі збільшеним апоптозом Т-лімфоцитів та вищою смертністю в моделі CLP на мишах.
У посмертному дослідженні Т-лімфоцити показали збільшення експресії PD‑1 та зменшення експресії рецепторів IL‑7 (IL‑7R або CD127) та CD28 на лімфоцитах [45].
Проліферація Т-лімфоцитів погіршується також при сепсисі [53, 85]. Нещодавно проведені дослідження показують, що експресія PD‑1 суттєво збільшується в периферичній крові на CD4+ і CD8+-лімфоцитах під час виділеної з крові інфекції Candida, пов’язаної з активацією CD8+ Т-лімфоцитів (збільшена експресія CD69), зменшення ко-стимулюючої експресії CD28 та IL‑7R [86].
В іншому проспективному клінічному дослідженні було виявлено, що в умовах прогресування сепсису T-лімфоцити збільшували експресію TIM‑3, LAG‑3, CTLA‑4 та зменшували експресію IL‑7R і секрецію IFN-γ при ex vivo симуляції поряд зі збільшенням експресії PD-L1 на дендритних клітинах [87]. Це дослідження доводить, що експресія інгібуючих рецепторів була порівняно низькою на початку сепсису, але суттєво збільшувалася, якщо сепсис прогресував [88].
N.Shubin та співавт. показали, що експресія BTLA була значно збільшена на циркулюючих CD4+ Т-лімфоцитах та В-лімфоцитах у септичних мишей, що було пов’язано зі зниженням кількості цих клітин [89]. Крім того, у пацієнтів у критичному стані із сепсисом збільшена експресія BTLA на циркулюючих CD4+ Т-лімфоцитах (>80%) корелювала зі збільшеною чутливістю до госпітальних інфекцій [89].
Відомо, що вентилятор-асоційована пневмонія є однією з найбільш розповсюджених госпітальних інфекцій при сепсисі. Дослідження Boomer та співавт. показало збільшення експресії PD-L1 та HVEM (HVEM-ліганд для BTLA) в легенях септичних хворих посмертне порівняно з контрольною групою [45].
Ці результати підтверджують гіпотезу, згідно з якою у септичних пацієнтів збільшена чутливість до вторинних інфекцій частково внаслідок збільшеної експресії інгібуючих лігандів на паренхімі легень, які забезпечують низькорівневу регуляцію функції Т-лімфоцитів [3].
Виснаження Т-лімфоцитів може також бути підсилено розчинними медіаторами, такими як IL‑10 та трансформуючий фактор росту-β (TGF-β) [90]. Високе співвідношення IL‑10 до TNF-α, як показали дослідження, корелювало зі зростанням летальності в пацієнтів із сепсисом та індукцією IL‑10 у ранній стадії сепсису [91, 92].
IL‑10 інгібував продукцію прозапальних цитокінів, таких як TNF-α, IL‑1, IL‑6, IFN-γ, та GM-CSF (моноцит-гранулоцит колонієстимулюючий фактор) і негативно впливав на відповідь імуноцитів [93].
Численні клінічні дослідження показали загальну депресію продукції цитокінів під час сепсису, хоча функціональна важливість цього спостереження залишається невизначеною [94-96].
IL‑10 може продукуватися великою кількістю типів клітин, з-серед яких основні продуценти – регулюючі Т-лімфоцити (Tregs). Tregs визначаються за допомогою цитофлуометрії як із фенотипом CD4+CD25+FoxP3+, так і з фенотипом CD4+CD25+CD127. Кількість їх збільшується під час підгострої фази сепсису. Зростання кількості Tregs під час сепсису корелює з виснаженням CD4+ T-лімфоцитів [97].
Tregs здатні погіршувати функції ефекторних Т-лімфоцитів, викликаючи супресію клітинної проліферації, секреції цитокінів, та індукувати апоптоз [98]. Проведені дослідження також демонструють взаємодію між Tregs та PD‑1, тому що одночасне виснаження Tregs та блокада PD‑1 викликали синергічне покращення вірусного контролю і реверсування виснаження CD8+ Т-лімфоцитів [99].
TGF-β є ще одним протизапальним цитокіном, який погіршує функції Т-лімфоцитів щодо продукції IL‑2 та проліферації клітин і викликає розвиток Tregs [100, 101]. Рівні TGF-β у сироватці крові були збільшені в пацієнтів у ранній стадії сепсису [102, 103]. Преклінічні дослідження також демонструють підвищення рівня TGF-β під час експериментального сепсису [104, 105].
TGF-β негативно впливав на проліферацію Т-лімфоцитів та погіршував викликану T-клітинами імунну відповідь у легенях [106]. А. Ayala та співавт. показали, що збільшення рівня TGF-β корелювало з пригніченням функцій спленоцитів (продукція IL‑2) у моделі CLP сепсису, а лікування моноклональними антитілами проти TGF-β зменшувало дисфункцію спленоцитів [104].
Саме тому протизапальні цитокіни, такі як IL‑10 та TGF-β, можуть сприяти індукованому погіршенню функції Т-лімфоцитів при сепсисі. Як було зазначено вище, збільшена експресія інгібуючих рецепторів на Т-лімфоцитах збільшувала число Treg та загальну депресію цитокінів із трансформацією в протизапальний фенотип та схиляла баланс на користь імуносупресії при сепсисі [3, 38].
Далі буде.
Біль, знеболення та інтенсивна терапія,
2018, № 3.
Тематичний номер «Хірургія, Ортопедія, Травматологія, Інтенсивна терапія» № 3 (33), вересень 2018 р.