


31 грудня, 2018
Артеріальна гіпертензія у дітей та підлітків

Артеріальна гіпертензія (АГ) – патологічний стан, який супроводжується постійним або періодичним підвищенням артеріального тиску (АТ) порівняно з віковою нормою. Значна поширеність цієї хвороби, прогресуюче зростання захворюваності серед дітей та підлітків, асоціація з ризиком ураження органів-мішеней, раннім розвитком атеросклерозу та його ускладненнями зумовлюють важливість проблеми АГ як у дорослій, так і в дитячій кардіології [1, 2].
Дослідження останніх десятиліть показали, що початок АГ у дитячому віці – один з основних факторів ризику несприятливих кардіологічних подій у подальшому житті. З метою змінити негативну динаміку збільшення частоти ускладнень захворювання і покращити діагностику, оцінку та лікування АГ у педіатричних пацієнтів Європейське товариство гіпертензії (ESH, 2016) та Американська академія педіатрії (AAP, 2017) опублікували оновлені рекомендації з профілактики і лікування високого АТ у дітей [3, 4]. Незважаючи на відмінності цих рекомендацій стосовно інтерпретації показників АТ, які характерні для різних ступенів захворювання, вони свідчать про значний прогрес у діагностиці цієї патології у дітей та націлюють фахівців на дотримання чіткого алгоритму [5, 6].
Епідеміологія
Поширеність АГ серед дітей та підлітків, згідно з даними популяційних досліджень, у різних країнах варіює від 5 до 14%. Причому серед учнів старших класів цей показник становить 18% [7-10], а серед хлопчиків-підлітків сягає 25,1% [11].
Згідно з результатами I етапу реалізації науково-практичної програми «Вивчення епідеміології первинної артеріальної гіпертензії та метаболічного синдрому у дітей та підлітків», ініційованої Асоціацією педіатрів України, підвищений АТ спостерігається у 10-15% дітей (у 1,5-2,5 раза частіше серед дітей з надлишковою масою тіла).
Етіологія та патогенез первинної АГ
В етіологічній структурі первинної АГ (ПАГ) виділяють фактори ризику, які є тригерами захворювання (табл. 1)..jpg)
Найбільш вагомий фактор ризику розвитку ПАГ у дітей – збільшення індексу маси тіла (ІМТ). За оцінками Н.Н. Каладзе та співавт. (2014), 40,2% підлітків віком 12-17 років із ПАГ мають надлишкову масу тіла. З іншого боку, встановлено, що у дітей з надлишковою масою тіла та ожирінням ризик розвитку АГ підвищується у 2-3 рази [11, 12].
У дітей із цією патологією часто виявляють різні метаболічні порушення. Так, незалежно від форми ПАГ (стабільна/лабільна), а також на ранніх стадіях її формування (гіпертензія «білого халата») у педіатричних пацієнтів спостерігається гіперурикемія [13, 14], зниження рівня ліпопротеїдів низької щільності та підвищення концентрації фактора Віллебранда [15]. У ході одного з досліджень у 71,1% дітей зі стабільною формою ПАГ та у 51,2% із лабільною формою ПАГ виявлено протромботичні порушення гемостазу [16].
Серед факторів ризику розвитку ПАГ у дітей вагоме значення мають також гіподинамія або, навпаки, неконтрольовані заняття спортом, активне і пасивне куріння, низька маса тіла при народженні та штучне вигодовування.
За даними Н.Н. Каладзе та співавт. (2014), 31,7% підлітків віком 12-17 років із ПАГ є рухливими, виконують ранкову гімнастику і займаються спортом, 15,2% проводять менше 2 год за комп’ютером і переглядом телепередач. Натомість 68,3% дітей ведуть пасивний спосіб життя, до 5 год і більше проводять за комп’ютером.
Враховуючи той факт, що ПАГ у дітей формується переважно у препубертатному і пубертатному віці, вагому роль у патогенезі захворювання відіграють порушення адаптації, які можуть розвиватись у період статевого дозрівання.
Як у вітчизняній, так і у світовій науковій літературі ПАГ розглядають як одну з «хвороб адаптації». Згідно з теорією адаптації пусковим механізмом розвитку цієї групи захворювань є неадекватна реакція організму на стресові фактори. У публікації F. Sparrenberger та співавт. (2009) на основі результатів метааналізу даних обстеження 52049 осіб показано, що хронічний стрес і насамперед порушена адаптивна реакція на нього є ймовірними причинами стійкого підвищення АТ.
У науковій літературі останніх років набув поширення термін «стрес-індукована гіпертензія» [17]. В експерименті показано, що у новонароджених щурів, які перенесли неонатальний стрес, у подальшому розвиваються гіпертрофія серця, активація ренін-ангіотензинової системи (РАС) та дисбаланс рецепторного апарату з переважанням експресії рецепторів ангіотензину I [18].
Згідно з теорією загального адаптаційного синдрому у відповідь на стресовий фактор послідовно залучаються дві програми адаптації – кататоксична і синтоксична. Кататоксична програма адаптації реалізується через адренергічну і гіпоталамо-гіпофізарно-наднирникову (стрес-реалізуючі) системи, а синтоксична – через ГАМК-ергічну та гіпоталамо-гіпофізарно-гонадну (стрес-лімітуючі) системи. При цьому доведено значущість певної послідовності залучення у патогенез ПАГ кататоксичної та синтоксичної програм адаптації.
На першому етапі стресової відповіді, який характерний для початку захворювання, активно залучається кататоксична програма, що реалізується через адренергічну та гіпоталамо-гіпофізарно-наднирникову системи. При цьому змінюється співвідношення норадреналіну/адреналіну і підвищується рівень кортизолу у плазмі крові. Припускають, що підвищення рівня кортизолу – це захисна реакція організму у відповідь на системне запалення. Кортизол у цьому випадку чинить протизапальний та протиалергічний ефекти, і зміна його рівня чітко корелює з іншими факторами неспецифічної запальної відповіді.
Як відомо, кортизол регулює процес реабсорбції натрію. Його низький рівень зумовлює затримку натрію, внаслідок цього може розвинутись АГ. Кортизол розглядають також як основний фактор формування інсулінорезистентності. Адже відомо, що інсулінорезистентність за своєю суттю є фізіологічною адаптивною реакцією, спрямованою на мобілізацію і перерозподіл потоків енергетичних субстратів (глюкози та вільних жирних кислот) між органами і тканинами в умовах стресу. Це покладено в основу глюкокортикоїд-метаболічної теорії цукрового діабету (ЦД) 2 типу, згідно з якою підвищення рівня кортизолу у таких пацієнтів спричиняє перехід фізіологічної інсулінорезистентності у патологічну.
Згідно з теорією загального адаптаційного синдрому тривала активація стрес-реалізуючих систем в умовах нормального гомеостазу призводить до компенсаторного підвищення активності стрес-лімітуючих систем адаптації, що реалізуються через ГАМК-ергічну та гіпоталамо-гіпофізарно-гонадну системи. До стрес-лімітуючих систем також належать серотонінергічна, антиоксидантна, система опіоїдних та інших регуляторних пептидів. Стрес-лімітуючі системи обмежують і модулюють активність стрес-реалізуючих систем, запобігаючи пошкоджуючій дії надмірної активації.
Компоненти стрес-реалізуючої системи – катехоламіни, адренокортикотропний гормон, кортизол, глюкагон, кортиколіберин, естрогени, компоненти згортальної системи крові, тромбоксан, лімфоцити СD8, оксидантна система, ангіотензин II, інтерлейкіни (IL) 1, 4, 6, 10, простагландин F, дофамін, фактор некрозу пухлини, Т2-хелпери, нейтрофільні лейкоцити, натрій, залізо, мідь, кальцій та ін. Компоненти стрес-лімітуючої системи – ацетилхолін, мелатонін, соматоліберин, соматотропний гормон, інсулін, прогестерон, компоненти протизгортальної системи крові, лімфоцити СD3, СD16, СD20, антиоксидантна система, оксид азоту, серотонін, простагландин Е1 та Е2, простациклін, натрійуретичні пептиди, інтерферон-γ, IL‑2 та IL‑12, гамма-аміномасляна кислота (ГАМК), гліцин, енкефаліни, β-ендорфін, Т1-хелпери, моноцити, еозинофіли, калій, магній, цинк, селен та ін.
Основним компонентом стрес-лімітуючої системи організму вважають гормон епіфізу мелатонін. Саме мелатонін відповідає за запуск процесу статевого дозрівання. У препубертатному періоді його рівень фізіологічно різко знижується, що зумовлює підвищення активності передньої долі гіпофіза, яке є необхідним для статевого дозрівання. Внаслідок зменшення гальмівного впливу мелатоніну посилюється дія вазоконстрикторних факторів.
У разі недостатньої активації стрес-лімітуючих систем розвиваються надмірна активація стрес-реалізуючих систем та порушення адаптації, що можуть спричинити АГ.
Слід зазначити, що у результаті підвищення активності як стрес-реалізуючих, так і стрес-лімітуючих систем організму розвивається стан, який характеризується стійкістю до сильного пошкоджуючого впливу. Як вказує Г. Сельє, підвищення резистентності досягається «дорогою ціною», оскільки супроводжується великими енерговитратами. У центральній нервовій системі (ЦНС) розвивається гальмування – і сильний подразник вже сприймається як слабкий або помірної сили.
Наявність фази резистентності при АГ можна припустити на підставі частої відсутності скарг у дітей із досить високим АТ та існування прихованої АГ. Приблизно у половини підлітків із ПАГ підвищений АТ вперше виявляють під час профілактичного огляду, що свідчить про малосимптомний перебіг захворювання.
За результатами досліджень за участю дорослих середнього віку встановлено, що перші 5-10 років від початку захворювання організм людини адаптується до підвищеного АТ, проте зі збільшенням тривалості гіпертензії активність адаптаційних процесів знижується.
ПАГ формується під дією факторів ризику за наявності певних індивідуальних особливостей організму (вегетативної реактивності, функціонування баро- та хеморецепторних механізмів контролю АТ, ренін-ангіотензин-альдостеронової системи, функції ендотелію, гомеостатичної функції нирок та ін.).
Як правило, АГ розвивається на тлі гіперактивності симпатичної вегетативної нервової системи (СВНС). Висока активність СВНС призводить до збільшення серцевого викиду та периферичної вазоконстрикції, внаслідок чого підвищується АТ.
Існують механізми контролю та обмеження підвищення АТ, до яких належать аортокаротидний барорефлекс (контроль АТ) та кардіопульмональний барорефлекс (контроль серцевого викиду). Вважається, що у патогенезі хронічного підвищення АТ певну роль відіграє їх генетично зумовлена дисфункція, оскільки у разі їх нормального функціонування відбувається інгібування довготривалої гіперактивації СВНС.
Симпатикотонія також розвивається при синдромі обструктивного апное уві сні. Епізоди гіпоксії у таких пацієнтів призводять до активації каротидних хеморецепторів, що проявляється не тільки підвищенням АТ уночі, а й зміною порогу чутливості цих рецепторів вдень. Тривале використання протягом ночі приладів, які створюють позитивний тиск у дихальних шляхах, забезпечує редукцію гіперсимпатикотонії та зниження АТ.
Потужним активатором СВНС є ендогенний нікотин. Хоча викурювання однієї цигарки призводить, як правило, до короткочасного підвищення АТ, у активних курців може розвинутися хронічна АГ.
Дія СВНС на тонус периферичних судин, частоту спонтанної активації синусового вузла та скоротливість міокарда серця є механізмом короткотривалої регуляції АТ. Причиною стабільної довготривалої АГ вважається дисфункція ренін-ангіотензин-альдостеронової системи (РААС). Проте існує взаємозв’язок між СВНС та функцією нирок, а саме з певними механізмами, які можуть бути залучені у патогенез АГ. Симпатичні імпульси є потужним стимулом для вивільнення у кровоток реніну через активацію β1-адренорецепторів юкстагломерулярного апарату нирок, а також для посилення реабсорбції натрію через активацію α1-адренорецепторів, які контролюють Na+-K+-АТФазу збірних трубочок. ПАГ може починатися з гіперактивації СВНС (початкова стадія – лабільна АГ) із подальшим залученням РААС, системи реабсорбції солей та води в нирках, що призводить до прогресування стабільної АГ.
Одна з основних патогенетичних ланок АГ – це спадкове або набуте порушення екскреції натрію нирками. Тимчасова затримка натрію і, як наслідок, води призводить до збільшення об’єму циркулюючої крові, що, у свою чергу, стимулює збільшення серцевого викиду та периферичну вазоконстрикцію.
Такий фактор, як низька маса тіла при народженні, також пов’язаний із порушенням нефрогенезу та розвитком гіпертензії, асоційованої з хлоридом натрію.
РААС відіграє провідну роль у розвитку та прогресуванні хронічної ПАГ. Ангіотензиноген, який синтезує печінка, під впливом реніну, продукованого юкстагломерулярним апаратом нирок, перетворюється на ангіотензин І. Ангіотензинперетворюючий фермент, який переважно міститься у судинах легень, меншою мірою – у судинах великого кола кровообігу, розщеплює ангіотензин І до ангіотензину ІІ. Останній чинить декілька важливих ефектів: підвищує тонус СВНС; діє безпосередньо на канальці нефрона та опосередковано через збільшення продукції альдостерону, стимулюючи реабсорбцію натрію та пасивну затримку води, а також екскрецію калію; зумовлює периферичну артеріолоконстрикцію; стимулює синтез антидіуретичного гормону гіпофізом, що також збільшує затримку води у збірних трубочках. Ці ефекти у сукупності зумовлюють затримку натрію та води і, як наслідок, збільшення об’єму циркулюючої крові та серцевого викиду; периферичну вазоконстрикцію; ремоделювання міокарда лівого шлуночка (ЛШ), гіпертрофію медії судин серця.
Важливу роль у розвитку АГ відіграє ендотелій артерій середнього та дрібного калібру, який бере участь у регуляції тонусу та проникності судин, підтриманні нормального співвідношення процесів про- та антикоагуляції, контролі адгезії та міграції імунних клітин крізь судинну стінку. Ендотеліальна активація проатерогенними факторами призводить до порушення усіх перелічених функцій, у тому числі до переважання синтезу вазоконстрикторних медіаторів (ендотеліну‑1) над синтезом вазодилататорних (оксиду азоту), внаслідок чого виникає периферичний вазоспазм і підвищується АТ.
Отже, з одного боку, проатерогенні фактори (куріння, ожиріння та дисліпідемія, гіперхолестеринемія у сімейному анамнезі, ЦД та ін.) зумовлюють персистуючу ендотеліальну дисфункцію, яка з часом може призвести до розвитку АГ, з іншого – саме АГ є проатерогенним фактором, що зумовлює порушення функції ендотелію. Таким чином замикається хибне коло – самопідтримання АГ через персистуючу ендотеліальну дисфункцію.
У випадку тривалої АГ місцеві судинні нейрогуморальні механізми призводять до ремоделювання стінки артерій за рахунок гіпертрофії медії. При цьому внаслідок зменшення просвіту дрібних судин відбувається стійке збільшення їх периферичного опору, після чого підвищується АТ, переважно за рахунок діастолічного АТ (ДАТ). Що стосується змін у магістральних артеріях, то гіпертрофія їх медії здебільшого є ексцентричною, що не впливає на внутрішній просвіт судини, проте значно збільшує жорсткість її стінки. Ізольовано цей механізм призводить до розвитку систолічної гіпертензії або поступової нормалізації ДАТ у пацієнтів із змішаним систолічно-діастолічним варіантом АГ через прогресуюче зниження здатності жорсткої стінки магістральних судин розтягуватися під впливом серцевого викиду і створювати діастолічний тиск.
Етіологія та патогенез вторинної АГ
Етіологічними факторами вторинної АГ є як спадкові, так і набуті ураження органів і систем, які спричиняють підвищення АТ.
На особливу увагу заслуговують спадкові захворювання, що супроводжуються підвищенням АТ, їх характеристика подана у таблиці 2 (В.Н. Коваленко, 2008). Усі відомі моногенні причини розвитку АГ характеризуються аномальним транспортом натрію в нирках, збільшенням об’єму циркулюючої крові та низьким рівнем реніну. Серед них ідентифіковано синдром Ліддла, синдром альдостеронізму (піддається корекції глюкокортикоїдами), синдром уявного надлишку мінералокортикоїдів, синдром Гордона, синдром підвищеної чутливості мінералокортикоїдних рецепторів та гіпертензивні форми вродженої гіперплазії кори наднирників. Моногенні захворювання слід запідозрити у дітей з АГ із низьким рівнем реніну у плазмі крові за наявності у сімейному анамнезі тяжкої гіпертензії з раннім початком або рефрактерної гіпертензії. Гіпокаліємія зустрічається у більшості у дітей з АГ із низьким рівнем реніну (крім тих, які мають синдром Гордона).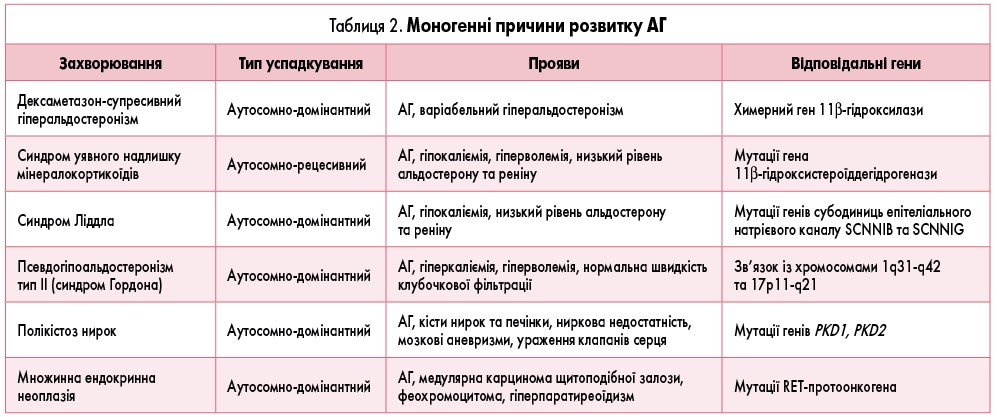
Ренопаренхіматозна АГ розвивається внаслідок ураження паренхіми нирок різного генезу (рубцювання при хронічному пієлонефриті, заміщення паренхіми при полікістозі нирок тощо). Патогенез захворювання полягає у зниженні секреції натрію та води, внаслідок чого збільшується об’єм циркулюючої крові, серцевий викид, виникає периферичний вазоспазм. Окрім цього, у більшості випадків ураження паренхіми нирок підвищується синтез реніну і, як наслідок, ангіотензину ІІ. Роль останнього у патогенезі АГ описана вище.
Реноваскулярну АГ спричиняє гіпоперфузія нирок внаслідок стенозу ниркових артерій (васкуліту, тромбозу, фібромускулярної дисплазії, зовнішнього стиснення об’ємними утвореннями). У відповідь на ішемію нирки відбувається гіперсинтез реніну, що призводить до гіперпродукції ангіотензину ІІ та підвищення АТ.
Коарктація аорти. Патогенез АГ у цьому випадку включає три механізми. По-перше, механічна перешкода кровотоку в низхідній аорті призводить до підвищення АТ проксимальніше місця коарктації. Саме тому типовою ознакою цієї патології є підвищення АТ при вимірюванні на плечі та його зниження при вимірюванні на підколінних артеріях. Якщо коарктація локалізується між лівою загальною сонною та лівою підключичною артеріями, то підвищення АТ реєструють на правій кінцівці, тоді як на інших трьох кінцівках – зниження АТ. По-друге, з часом через збільшення жорсткості дуги аорти її барорецептори перестають реагувати на АГ, таким чином виключається барорефлекс, який у нормальних умовах обмежує підвищення АТ. По-третє, зниження кровонаповнення ниркових артерій (оскільки місце їх відходження знаходиться дистальніше ділянки коарктації) призводить до гіперпродукції реніну та ангіотензину ІІ.
Ендокринні захворювання. АГ на тлі захворювань цієї групи розвивається внаслідок порушень гормонального фону. Так, для феохромоцитоми характерна гіперпродукція катехоламінів, що спричиняє периферичний вазоспазм, тахікардію та збільшення серцевого викиду, що призводить до розвитку АГ. У випадку гіперфункції кори наднирників (первинний гіперальдостеронізм, хвороба чи синдром Іценка – Кушинга) посилення синтезу альдостерону або кортизолу призводить до підвищення реабсорбції натрію та води, збільшення об’єму циркулюючої крові та серцевого викиду і, як наслідок, до підвищення АТ. Гіпертиреоз характеризується збільшенням рівня тироксину та/або трийодтироніну у сироватці крові, що зумовлює підвищену чутливість тканин (у тому числі серця та судин) до дії катехоламінів та збільшення об’єму циркулюючої крові.
Патогенез ураження органів-мішеней. Основними ускладненнями АГ є гіпертрофія міокарда ЛШ, гіпертензивна ретинопатія, гіпертензивна нефропатія, ураження церебральних судин.
Доведено, що незалежно від причини підвищений АТ у дітей призводить до зміни геометрії ЛШ і збільшення товщини стінок серця, причому маса серця збільшується вже на ранній стадії АГ. У дітей спостерігається збільшення індексу маси міокарда ЛШ на 0,14 одиниць на кожні 10 одиниць систолічного АТ (САТ) за даними добового моніторингу АТ (ДМАТ) [19].
Залежно від патогенезу АГ виділяють різні типи гіпертрофії міокарда ЛШ. У випадку переважного збільшення постнавантаження (гіперсимпатикотонічний артеріолоспазм, реноваскулярна гіпертензія, коарктація аорти) розвивається концентрична гіпертрофія ЛШ, що характеризується потовщенням стінок без зміни або зі зменшенням розмірів його порожнини. При АГ зі збільшеним пост- та пренавантаженням (ренопаренхіматозна АГ, первинний гіперальдостеронізм) гіпертрофія є ексцентричною – з потовщенням стінок та розширенням порожнини ЛШ. У будь-якому випадку описане ремоделювання міокарда призводить до збільшення його жорсткості та діастолічної дисфункції ЛШ. Окрім цього, гіпертрофія ЛШ може бути причиною розвитку відносної ішемії міокарда (через відсутність адекватного ступеня гіпертрофії збільшення коронарного кровотоку), дисметаболічних змін та міокардіосклерозу, внаслідок чого з часом до діастолічної може приєднуватися систолічна дисфункція ЛШ із виникненням у подальшому хронічної серцевої недостатності (ХСН).
Ураження судин, найбільш характерним з яких є церебральна ангіопатія, розвивається за кількома клінічними сценаріями. При гіпертензивному кризі (ГК) різко збільшується проникність дрібних судин головного мозку, внаслідок чого масивний транссудат рідини може призвести до його набряку. Проникність судин може збільшитися настільки, що шляхом діапедезу виникають дрібновогнищеві крововиливи вздовж ходу церебральних судин. Нарешті при ГК можливий розрив судин середнього калібру, що може спричинити розвиток геморрагічного інсульту або субарахноїдальний крововилив. Останнє ускладнення ГК виникає внаслідок тривалого впливу підвищеного АТ на стінки церебральних судин, що призводить до формування мікроаневризм, локального витончення чи розвитку атеросклеротичного ураження.
Навіть за відсутності ГК тривала АГ спричиняє поступове ремоделювання церебральних артеріол із розвитком концентричної гіпертрофії медії та стійким зменшенням внутрішнього їх діаметра. Проградієнтне звуження дрібних судин поступово призводить до відносної ішемії церебральних тканин, прогресуючої дисциркуляторної енцефалопатії з атрофією речовини мозку та деменції, яка більш характерна для людей похилого віку з тривалою АГ. У дітей із цим захворюванням зміни у церебральних судинах менш виражені порівняно з дорослими, проте і в них спостерігається зниження когнітивних функцій, які покращуються на тлі лікування антигіпертензивними препаратами [20, 21].
Гіпертензивна нефропатія виникає внаслідок гіпертрофії гладеньком’язових клітин дрібних судин та некрозу стінки ниркових капілярів на тлі стійкого та значного підвищення АТ. Це призводить до поступової ішемічної атрофії канальців нирок (меншою мірою – клубочків), що з часом у випадку тривалої неконтрольованої АГ може спричинити розвиток хронічної ниркової недостатності.
Нирки у дітей з АГ функціонують у режимі гіперфільтрації та відносного, а у половини дітей – абсолютного, порушення екскреції.
Гіпертензивна ретинопатія характеризується крововиливами або інфарктами сітківки, набряком диска зорового нерва або помірно прогресуючими змінами у вигляді потоншання судинної стінки артеріол внаслідок гіпертрофії їх медії та артеріосклерозу. Останній варіант, як правило, не супроводжується клінічними проявами з боку органів зору, проте є індикатором недостатнього контролю захворювання.
Ангіопатія сітківки спостерігається більш ніж у половини підлітків із ПАГ (61,8%). Ступінь вираженості ангіопатії різний – від ізольованого звуження артерій (19,1%) або розширення вен (6,6%) до значного поєднаного ураження артеріальних і венозних судин (36%) (В.Г. Майданник та ін., 2014).
Діагностика АГ у дітей
Дослідження, проведені у країнах Європи та США, показали, що у дітей АГ правильно діагностують тільки у 13-26% випадків [22]. За результатами мультицентрового дослідження [22, 23] тільки 25% педіатрів регулярно вимірюють АТ у своїх пацієнтів, у той час як 71% вимірювали його тільки за наявності у дитини факторів ризику розвитку АГ, при цьому 47% педіатрів недооцінили категорію АТ у своїх пацієнтів.
 Вимірювання АТ у дітей. Планове вимірювання АТ дитини під час кожного візиту до лікаря рекомендовано проводити з 3-річного віку. У молодших дітей офісну тонометрію проводять тільки за клінічної необхідності (перебування у відділенні інтенсивної терапії чи реанімації, наявність вродженої вади серця, захворювання нирок, внутрішньочерепної гіпертензії, прийом ліків, що підвищують АТ).
Вимірювання АТ у дітей. Планове вимірювання АТ дитини під час кожного візиту до лікаря рекомендовано проводити з 3-річного віку. У молодших дітей офісну тонометрію проводять тільки за клінічної необхідності (перебування у відділенні інтенсивної терапії чи реанімації, наявність вродженої вади серця, захворювання нирок, внутрішньочерепної гіпертензії, прийом ліків, що підвищують АТ).
Після того, як за результатами вимірювання АТ дитина оцінена як нормотензивна, перевірку необхідно здійснювати щороку.
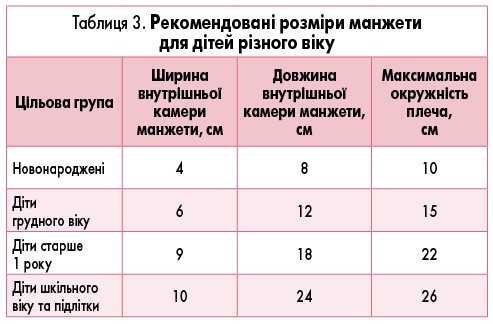
Під час вимірювання АТ важливо дотримуватися вимог щодо підбору манжети відповідного розміру залежно від віку дитини (табл. 3).
 У дітей вимірювання АТ проводять на правій руці у положенні сидячи або лежачи. Перед вимірюванням дитина має протягом 5 хв перебувати у спокої в сидячому/лежачому положенні, необхідно створити тиху і комфортну обстановку. Недопустимо проводити вимірювання АТ невдовзі після фізичного навантаження, прийому стимуляторів (наприклад, кофеїну) чи куріння впродовж останніх 30 хв, а також у шумному чи задушливому приміщенні, оскільки це може вплинути на його результати.
У дітей вимірювання АТ проводять на правій руці у положенні сидячи або лежачи. Перед вимірюванням дитина має протягом 5 хв перебувати у спокої в сидячому/лежачому положенні, необхідно створити тиху і комфортну обстановку. Недопустимо проводити вимірювання АТ невдовзі після фізичного навантаження, прийому стимуляторів (наприклад, кофеїну) чи куріння впродовж останніх 30 хв, а також у шумному чи задушливому приміщенні, оскільки це може вплинути на його результати.
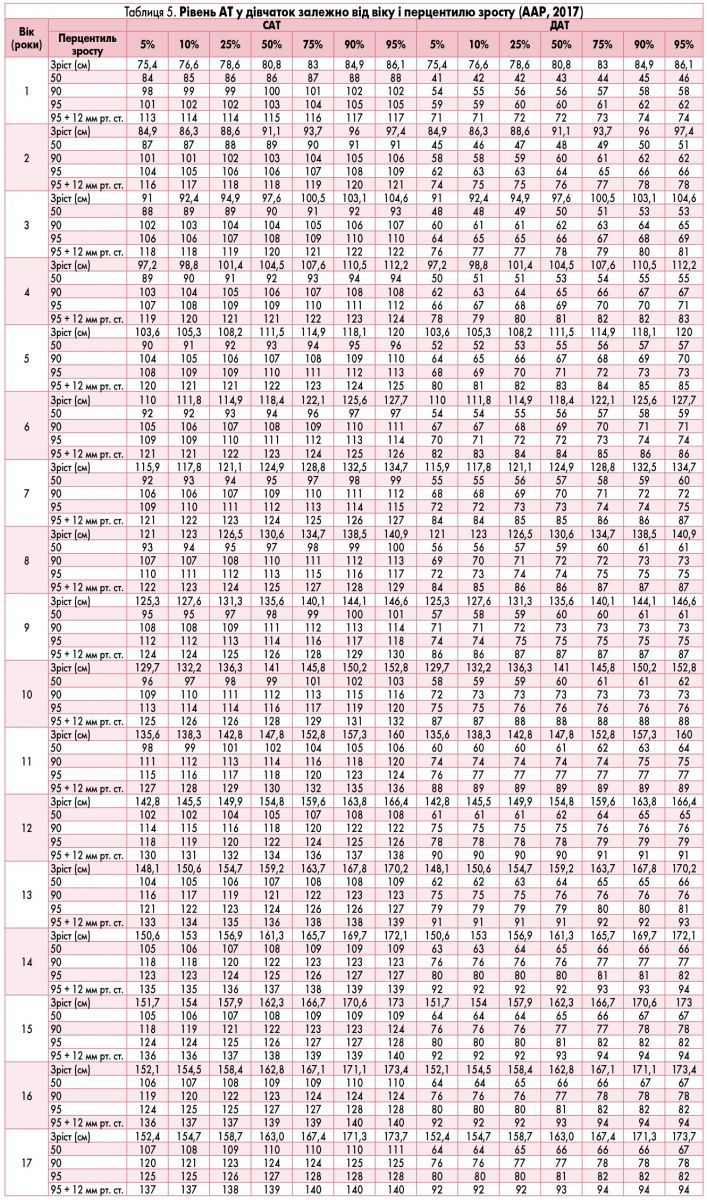
Показники САТ і діастолічного АТ (ДАТ) порівнюють із перцентильним розподілом нормативних значень згідно з перцентилем зросту для відповідного віку та статі (табл. 4 і 5) за алгоритмом, представленим на рис. 1.
Якщо САТ і ДАТ <90-го перцентилю, повторна тонометрія під час візиту не потрібна. Якщо САТ або ДАТ >90-го перцентилю, проводять два додаткові вимірювання з інтервалом 5 хв та розраховують середнє арифметичне значення АТ. Для оцінки АТ використовують середні значення САТ і ДАТ пацієнта, отримані під час трьох візитів з інтервалом 10-14 днів.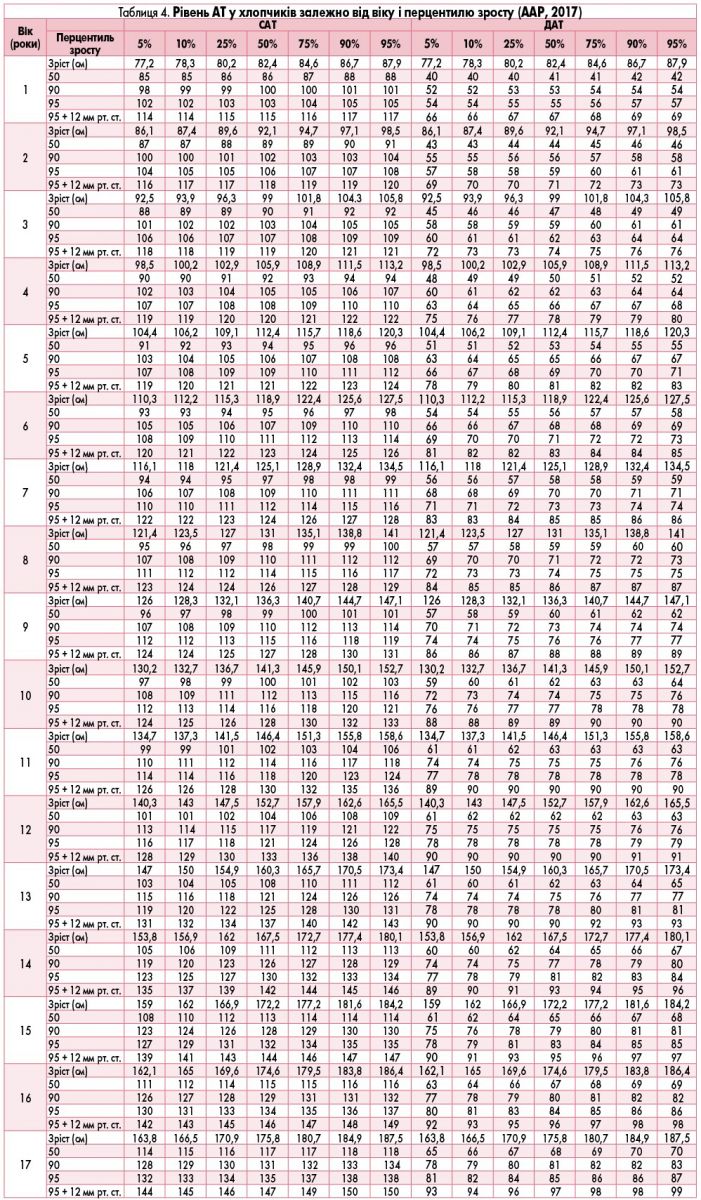
Добове моніторування АТ (ДМАТ). Цей метод передбачає використання портативних автоматичних цифрових тонометрів для отримання добового профілю АТ у пацієнта і розрахунку діагностично значимих похідних. Порівняно з офісним вимірюванням тиску ДМАТ дає можливість точно встановити ступінь АГ і, відповідно, визначити тактику ведення пацієнта.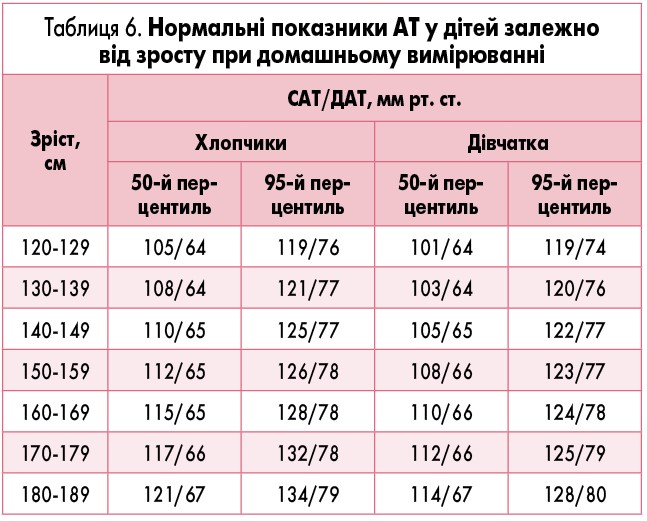
Домашнє вимірювання АТ. Останнім часом у дорослій та дитячій кардіології дедалі більшої ваги набуває домашня тонометрія [24]. Окремі дослідження показали, що цей метод має вищий ступінь відтворюваності порівняно з офісною тонометрією та не поступається за наведеною характеристикою ДМАТ. Переваги домашнього вимірювання АТ над офісним такі: комфортна атмосфера, виключення впливу на показники ефекту «білого халата», можливість отримання графіка тривалого щоденного моніторингу. Проте домашня тонометрія потребує від батьків або дитини навичок її проведення та унеможливлює контроль лікаря за дотриманням правил вимірювання АТ. Домашня тонометрія порівняно з ДМАТ дає можливість лише вибірково оцінити АТ, не дозволяє оцінити навантаження тиском та утруднює встановлення кореляції показників зі скаргами дитини, проте забезпечує тривалий моніторинг АТ у домашніх умовах, не створюючи дискомфорт для дитини через постійне автоматичне вимірювання АТ вночі.
Вимірювання АТ рекомендовано проводити вранці та ввечері двічі (та розраховувати середнє арифметичне значення АТ для кожного часу доби) протягом 6-7 днів. Для цього використовують напівавтоматичні або автоматичні тонометри. Для оцінки результатів вимірювання АТ можна використати дані, наведені у таблиці 6.
Термінологія та класифікація АГ
Нормальним АТ у дітей вважають рівні САТ і ДАТ ≥10-го та <90-го перцентилю розподілу нормативних показників залежно від віку, статі та зросту.
У дітей діагноз АГ встановлюють, якщо середні значення САТ та/або ДАТ ≥95-го перцентилю розподілу нормативних показників залежно від віку, статі та зросту, які отримані протягом ≥3 візитів (з інтервалом не менше 10-14 днів).
Високий нормальний АТ у дитини встановлюють, якщо середнє арифметичне значення САТ та/або ДАТ перебуває у діапазоні ≥90-го та <95-го перцентилю протягом ≥3 візитів.
Артеріальна гіпертензія
У таблиці 7 подано класифікацію ПАГ, прийняту на X Конгресі педіатрів України (2014 р.).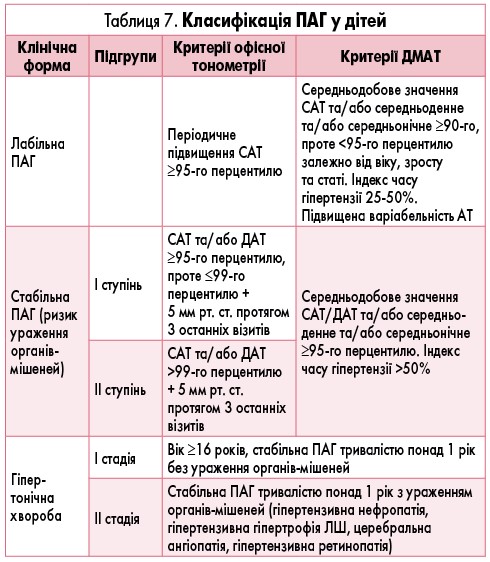
Якщо рівні САТ і ДАТ належать до різних категорій (наприклад, САТ – ІІ ступінь ПАГ, ДАТ – І ступінь ПАГ або САТ – І ступінь ПАГ, ДАТ – нормальний рівень), то ступінь ПАГ встановлюють за вищим із двох рівнів.
В останніх рекомендаціях ААР 2017 року [3] наголошується, що для підлітків віком від 13 років і старше незалежно від статі встановлення діагнозу АГ на підставі даних офісного вимірювання АТ не має ґрунтуватися на 95-му перцентилі. Рекомендовано використовувати абсолютне значення АТ, як у дорослих пацієнтів (табл. 8).
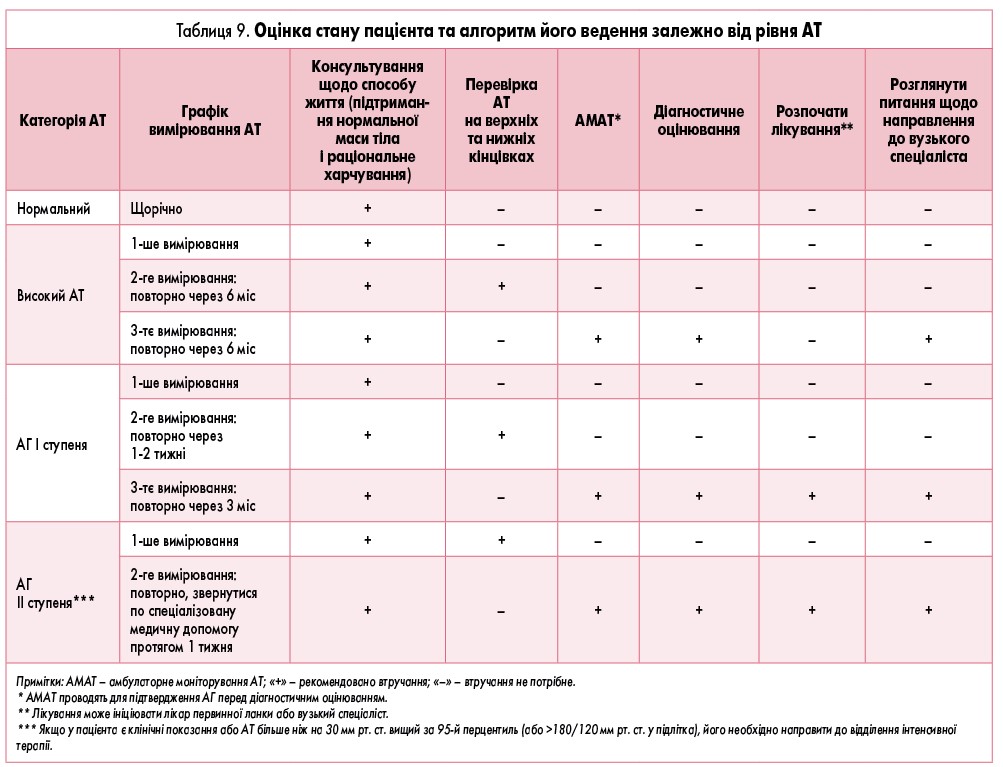
Якщо рівень АТ >90-го перцентилю, необхідно провести повторне вимірювання у термін, який залежить від того, до якої категорії належить дитина (табл. 9). У дітей з високим АТ інтервал між вимірюваннями становить 6 міс. Якщо при повторному вимірюванні знову визначається високий АТ, проводять 3-тє вимірювання ще через півроку, за результатами якого направляють дитину на амбулаторний моніторинг АТ (АМАТ) і на консультацію до вузьких спеціалістів.
На підставі результатів ДМАТ встановлюють гіпертензію «білого халата», якщо показники пацієнта відповідають діагностичним критеріям АГ за даними офісного вимірювання, при цьому середні показники АТ за даними ДМАТ <95-го перцентилю залежно від зросту, а індекс часу гіпертензії <25%. Для підтвердження цього діагнозу можна також використовувати домашнє вимірювання АТ. Гіпертензія «білого халата» є фактором ризику розвитку справжньої АГ.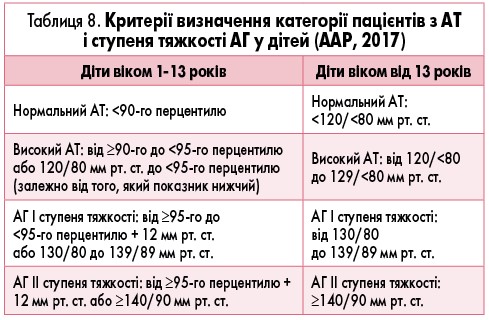
Виокремлюють також масковану гіпертензію, якщо показники пацієнта не відповідають критеріям АГ за даними офісного вимірювання, але перевищують середні показники АТ за даними ДМАТ 95-го перцентилю залежно від зросту та індексу часу гіпертензії >25%.
Оцінка стану органів-мішеней. При АГ органами-мішенями є нирки, сітківка ока, міокард ЛШ та магістральні судини (зокрема церебральні). Для виявлення гіпертензивної нефропатії рекомендовано провести визначення мікроальбумінурії (МАУ), оскільки цей тест є більш чутливим на ранніх стадіях ураження нирок порівняно з вимірюванням концентрації білку в добовій сечі. Необхідно також визначити рівень креатиніну сироватки крові та розрахувати швидкість клубочкової фільтрації (ШКФ) за формулою Шварца, яку використовують для оцінки функції нирок:
 ШКФ (мл/хв/1,73 м2) = (К × зріст (см) × 113) / рівень креатиніну сироватки крові (мкмоль/л),
ШКФ (мл/хв/1,73 м2) = (К × зріст (см) × 113) / рівень креатиніну сироватки крові (мкмоль/л),
де К=0,413 (виняток: у дітей віком до 5 років К=0,313; у хлопчиків старше 13 років К=0,616). Нормальна ШКФ >90 мл/хв/1,73 м2.
Для виявлення гіпертрофії міокарда ЛШ пацієнтам проводять електрокардіографію (ЕКГ) та двовимірну ехокардіографію (ЕхоКГ). Одним з ЕКГ-критеріїв є ознака Соколова – Лайона – сума амплітуд зубця S у відведенні V1 та зубця R у V5 або V6 (більша з них) >38 мм.
Масу міокарда ЛШ визначають за формулою, запропонованою R.B. Devereux та співавт. (1986) і рекомендованою для використання у дітей з АГ:
ММЛШ = 0,8 × [1,04 × (ТМШП + ТЗСЛШ + КДРЛШ)3 – (КДРЛШ)3] + 0,6,
де КДРЛШ – кінцеводіастолічний розмір лівого шлуночка, ТЗСЛШ – товщина задньої стінки ЛШ, ТМШП – товщина міжшлуночкової перетинки.
Індекс маси міокарда ЛШ (ІММ ЛШ) розраховують за формулою: ІММЛШ = ММЛШ / ріст 2,7.
Гіпертрофію ЛШ встановлюють, якщо ІММЛШ >95-го перцентилю для відповідного віку та статі (табл. 10).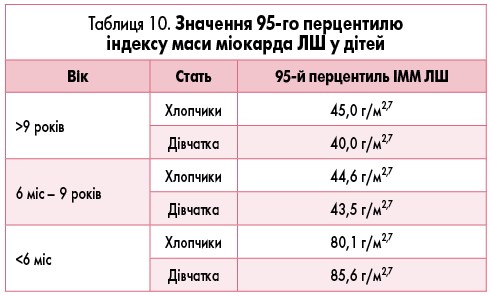
Як нормативні значення ІММЛШ можна також використовувати значення 95-го перцентилю залежно від віку і статі, які розраховані за рівнянням лінійної регресії, запропонованим P.R. Khoury та співавт. [25].
Для хлопчиків:
ІММЛШ95 = 77,5265 – вік × 15,8939 + вік1,5 × 5,2322 – вік2 × 0,4671 + 2,7380.
Для дівчаток:
ІММЛШ95 = 77,5265 – вік × 15,8939 + вік1,5 × 5,2322 – вік2 × 0,4671.
Для визначення типу гіпертрофії ЛШ (концентрична, ексцентрична) використовують показник відносної товщини стінки ЛШ (ВТСЛШ), який розраховують за формулою: ВТСЛШ = 2 × ТЗСЛШ/КДРЛШ.
Згідно з класифікацією R.M. Lang та співавт. [26], яка рекомендована Американським товариством ехокардіографії (ASE) та Європейською асоціацією ехокардіографії (Euroecho), виділяють такі типи ремоделювання: нормальна геометрія ЛШ (ІММЛШ не збільшений, ВТСЛШ <0,42); концентричне ремоделювання (нормальний ІММЛШ і ВТСЛШ >0,42); концентрична гіпертрофія ЛШ (збільшення ІММЛШ вище норми і ВТСЛШ >0,42); ексцентрична гіпертрофія ЛШ (збільшення ІММЛШ, ВТСЛШ <0,42).
Огляд офтальмологом очного дна дозволяє виявити гіпертензивну ретинопатію на початкових стадіях (звуження артерій сітківки) або тяжкого ступеня (геморагії, ексудати, набряк диска зорового нерва). Класифікація гіпертензивної ретинопатії за Wong та Mitchell представлена у таблиці 11 [27].
Клінічне обстеження дітей з АГ
Збір анамнезу та об’єктивне обстеження пацієнта з АГ мають на меті виявлення ознак вторинної гіпертензії, ураження органів-мішеней та наявності супутніх станів. Необхідно з’ясувати особливості сімейного анамнезу для встановлення можливих спадкових причин розвитку АГ: наявність у близьких родичів гіпертонічної хвороби, ішемічної хвороби серця чи інсульту, ЦД, дисліпідемії, ожиріння, спадкових захворювань нирок (полікістозної нирки), ендокринних захворювань (феохромоцитоми, хвороби чи синдрому Іценка – Кушинга та ін.). Анамнез життя пацієнта передбачає отримання інформації про масу тіла та гестаційний вік при народженні (недоношеність та низька маса тіла при народженні асоційовані з розвитком АГ), шкідливі звички (куріння, вживання алкоголю, кофеїну та інших психостимуляторів), наявність хронічних захворювань, які можуть бути пов’язані з розвитком АГ (хронічної хвороби нирок, ЦД, гіпертиреозу), синдрому обструктивного нічного апное. Важливе значення має оцінка клінічних ознак, які вказують на вторинну природу АГ (поліурія, полідипсія, ніктурія, гематурія, набряки, різке схуднення чи збільшення маси тіла).
Слід звернути увагу на симптоми, які характерні для ураження органів-мішеней: гіпертензивну церебральну ангіопатію / дисциркуляторну енцефалопатію (головний біль, носові кровотечі, запаморочення, випадок порушення мозкового кровообігу, парезу лицевого нерва, судомного нападу на тлі ГК), гіпертензивну ретинопатію (порушення зору), гіпертрофію ЛШ / серцеву недостатність (біль у ділянці серця, задишка, набряки кінцівок, гепатомегалія, асцит), гіпертензивну нефропатію (гематурія, набряки, олігоурія).
Фізикальне обстеження дитини з АГ обов’язково має включати: вимірювання зросту та маси тіла з розрахунком ІМТ, визначення окружності талії, пальпацію пульсу на ліктьових, стегнових та підколінних артеріях, вимірювання АТ на чотирьох кінцівках. Під час фізикального обстеження пацієнта з АГ слід зосередити увагу на тих ознаках, які можуть вказувати на етіологію АГ (табл. 12).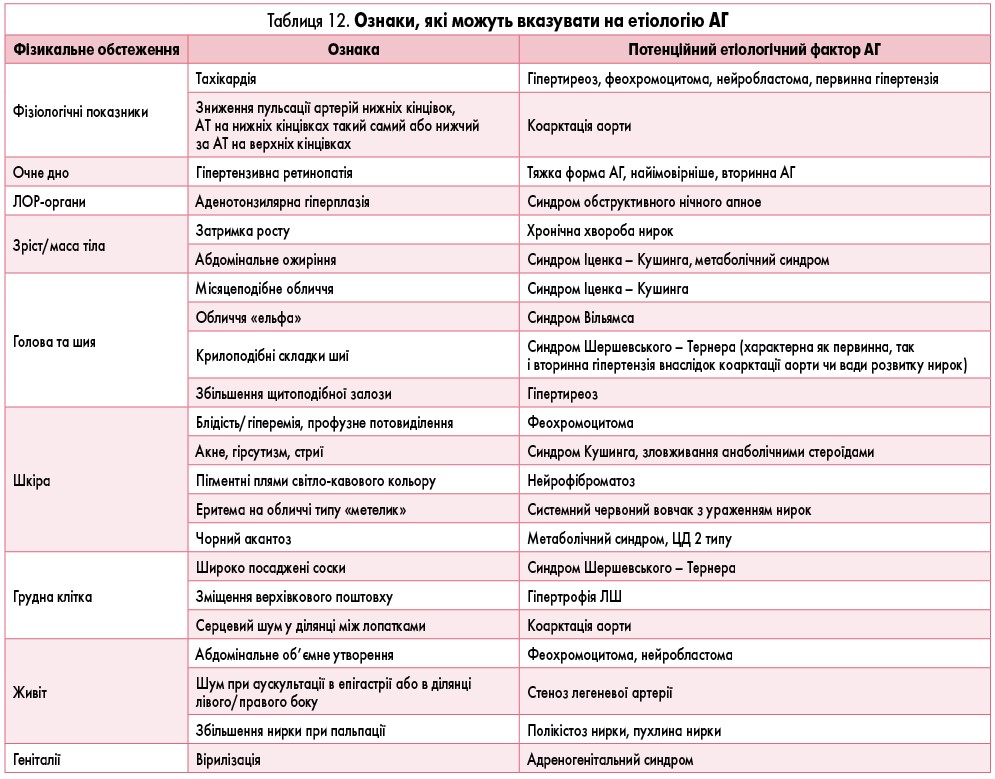
Лабораторні та додаткові інструментальні дослідження. Параклінічні дослідження проводять як для диференційної діагностики АГ, так і для виявлення ураження органів-мішеней. Рекомендований перелік досліджень у дітей з АГ наведено у таблиці 13.
Слід відзначити, що аналіз активності реніну плазми крові є інформативним методом для встановлення патогенетичного механізму АГ. Якщо активність реніну висока, РААС відіграє головну роль у патогенезі захворювання, тоді як низька активність реніну вказує на провідну роль затримки натрію та води. Якщо ж визначається нормальна активність реніну сироватки крові, у патогенез АГ залучені обидва механізми. Низька активність реніну має насторожити лікаря щодо можливих спадкових моногенних причин симптоматичної АГ, особливо за наявності сімейного анамнезу вказаних вище захворювань, високого або низького рівня калію чи альдостерону сироватки крові, ознак вірилізації чи гіпогонадизму. При цьому важливе діагностичне значення має альдостерон-ренінове співвідношення (ARR). Встановлено, що ARR >10 у дитини з АГ є показанням для генетичного тестування.
Диференційна діагностика АГ
Враховуючи розглянуті особливості клінічних, інструментальних і лабораторних досліджень у дітей з різними формами захворювання, можна сформувати алгоритм диференційної діагностики АГ (рис. 2).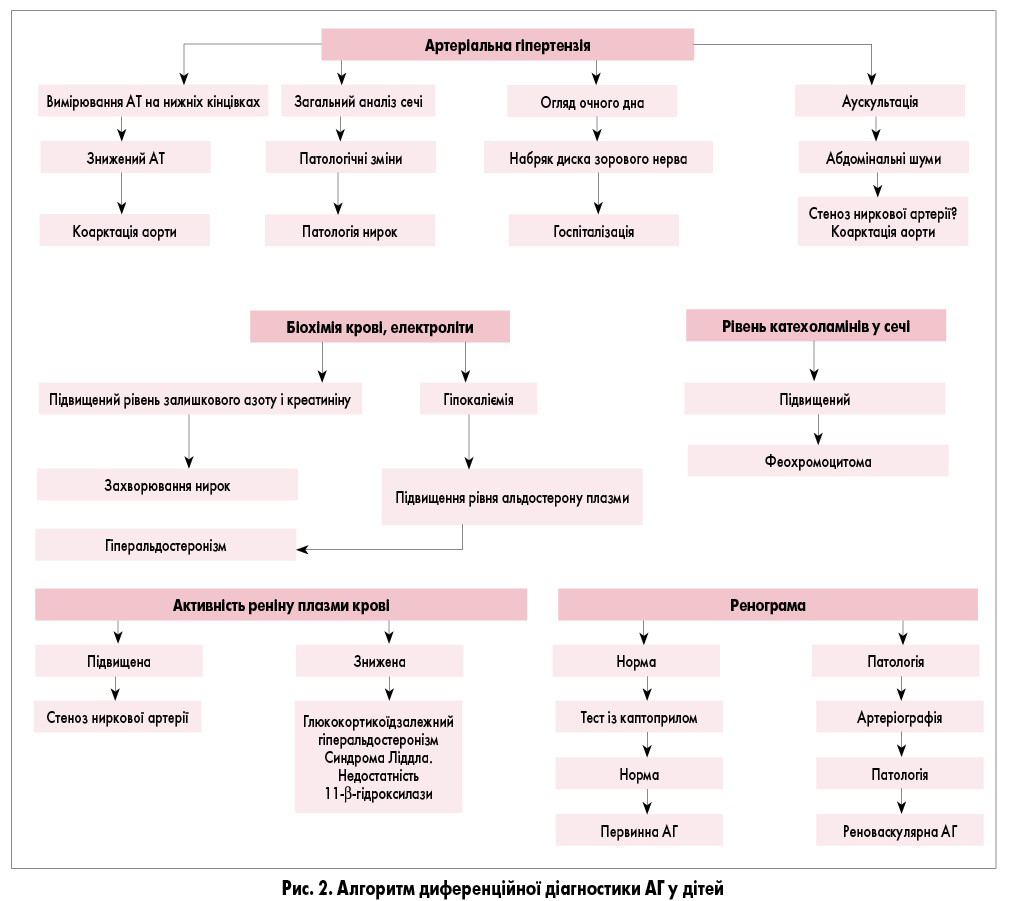
Лікування АГ
Лікування АГ у дітей передбачає комплекс заходів, серед яких на перший план виступають немедикаментозні методи: відмова від шкідливих звичок, корекція маси тіла, рекомендації щодо дієти, режиму дня та фізичних навантажень.
Якщо дитина має надлишкову масу тіла, її слід зменшити до ІМТ <85-го перцентилю зі швидкістю 1-2 кг за місяць.
Цільові показники АТ у дітей такі: <90-го перцентилю або <130/80 мм рт. ст. у підлітків віком ≥13 років.
Рекомендації щодо харчування дітей з АГ передбачають дотримання дієти DASH з переважанням фруктів та овочів, молочних продуктів із низьким вмістом жирів, продуктів із цільного зерна і бобових, обмеженням цукру і натрію (<2300 мг на добу).
Адекватне фізичне навантаження – важлива умова не тільки контролю маси тіла, а й обов’язкова складова немедикаментозного лікування дітей з АГ навіть без ожиріння. Короткочасна гіперсимпатикотонія та підвищення АТ внаслідок фізичного навантаження при регулярних заняттях спортом розвивають «тренованість» симпатичного відділу ВНС, яка проявляється зниженням рівня його базальної та стимульованої активності, що забезпечує профілактику АГ чи зниження АТ за її наявності. Як адекватне фізичне навантаження рекомендоване аеробне навантаження середньої та/або високої інтенсивності тривалістю 40 хв на день 3-5 днів на тиждень.
Прикладами фізичного навантаження середньої інтенсивності можуть бути: ходьба швидким кроком (3 км за 30 хв), танці у швидкому темпі (30 хв), гра в баскетбол (15-20 хв) або волейбол (45 хв), їзда на велосипеді (8 км за 30 хв).
Заборона брати участь у спортивних змаганнях поширюється тільки на дітей із неконтрольованою АГ ІІ ступеня. Рекомендовано обмежити «сидяче» проводження часу (відеоігри, перегляд телепередач) сумарно до 2 год на день. Обов’язковою є відмова від куріння та вживання алкоголю. Для модифікації способу життя, а саме харчової поведінки та фізичного навантаження, вкрай важливими є розуміння та підтримка оточення дитини, а також за необхідності – зміна традицій у родині, якщо вони суперечать поставленим терапевтичним цілям та перешкоджають реалізації програми їх досягнення.
Продовження читайте у наступному номері.
Тематичний номер «Педіатрія» № 4 (47) грудень 2018 р.