


31 травня, 2019
Новые механизмы развития неалкогольной жировой болезни печени и стратегия патогенетической терапии
 Неалкогольная жировая болезнь печени (НАЖБП) является наиболее распространенным заболеванием, от которого страдает около 25% взрослого населения во всем мире. НАЖБП – серьезная проблема общественного здравоохранения, связанная с эпидемией ожирения, нездоровыми диетическими моделями и распространением малоподвижного образа жизни.
Неалкогольная жировая болезнь печени (НАЖБП) является наиболее распространенным заболеванием, от которого страдает около 25% взрослого населения во всем мире. НАЖБП – серьезная проблема общественного здравоохранения, связанная с эпидемией ожирения, нездоровыми диетическими моделями и распространением малоподвижного образа жизни.
Быстро повышающаяся распространенность заболевания и его агрессивной формы – неалкогольного стеатогепатита (НАСГ) – требует новых терапевтических подходов, основанных на глубоком понимании факторов риска и патогенетических механизмов развития заболевания, с целью прекращения прогрессирования болезни до стадии фиброза или цирроза и рака печени. Новые данные свидетельствуют о том, что наблюдаемое в последнее время увеличение заболеваемости гепатоцеллюлярной карциномой обусловлено НАЖБП, особенно за рубежом [24]. Риск развития НАЖБП у женщин в репродуктивный период ниже по сравнению с мужчинами, однако с возрастом защитный эффект теряется, и распространенность НАЖБП сопоставима с таковой у мужчин.
Несмотря на то что в последние десятилетия были достигнуты значимые прорывы в отношении изучения патогенеза НАЖБП, заболевание по-прежнему остается сложным и мультисистемным, и понимание его механизмов все еще не полно.
В 2018 году были достигнуты существенные успехи в изучении факторов риска прогрессирующего заболевания печени при НАЖБП, включая генетические варианты и микробиоту кишечника (рис. 1, 2). Получены также обнадеживающие результаты применения лекарственных средств, воздействующих на метаболические пути, вовлеченные в прогрессирование повреждения печени.
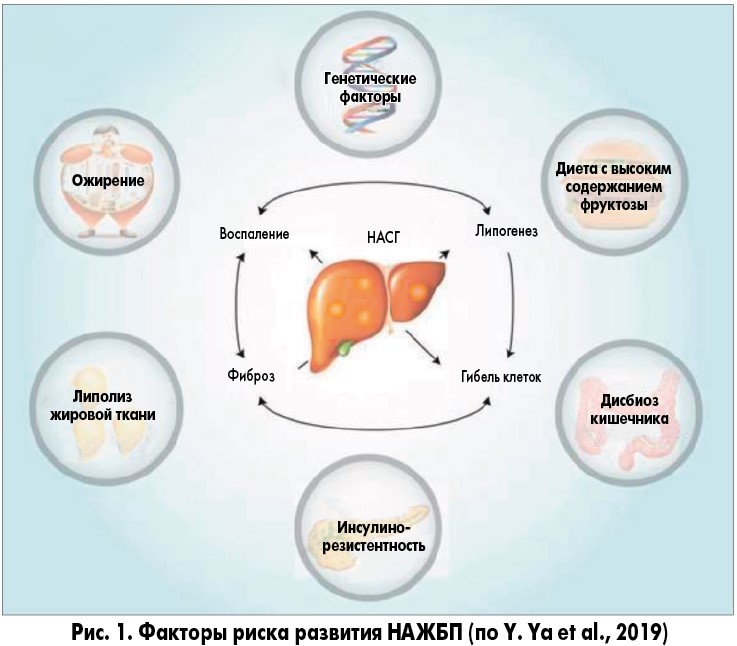
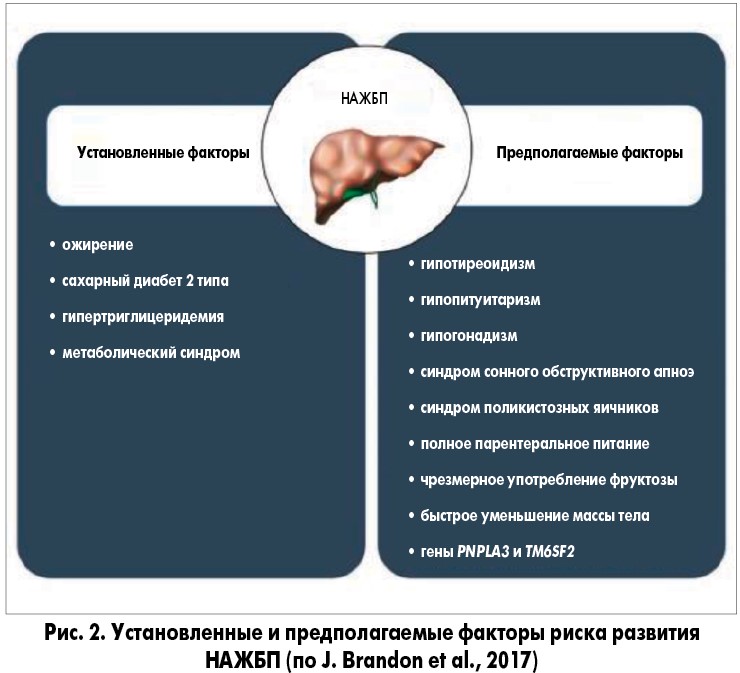
В настоящее время гипотеза «множественных ударов», которая подразумевает множество факторов, действующих параллельно и синергически у лиц с генетической предрасположенностью, является общепринятой точкой зрения для объяснения разных стадий развития НАЖБП, наблюдаемых клинически (рис. 3).
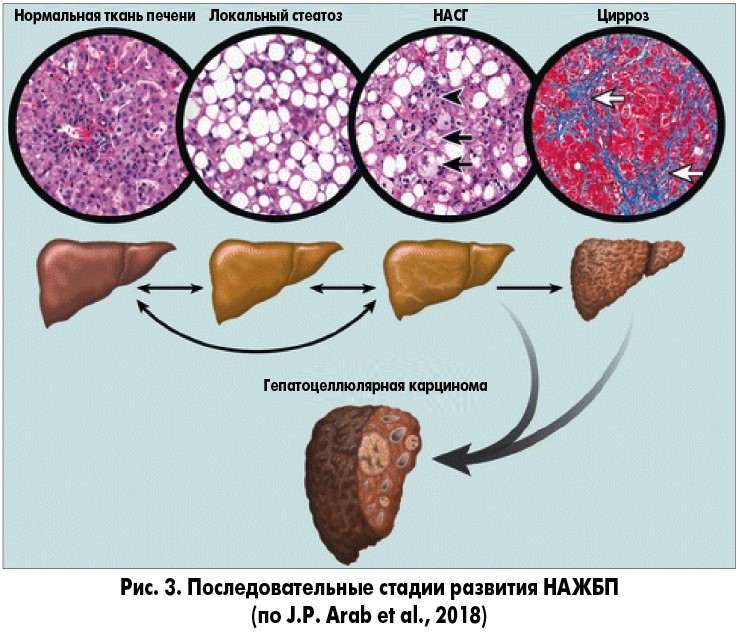
Новые концепции патогенеза, подтвержденные результатами клинических исследований, свидетельствуют о том, что стеатоз печени и НАСГ могут считаться изолированными, совершенно самостоятельными заболеваниями и не рассматриваться как последовательный процесс трансформации стеатоза в стеатогепатит. Однако эта точка зрения не является общепризнанной и требует дальнейшего изучения [2, 13].
Наследственные факторы и отличительные признаки некоторых этнических групп позволяют предположить, что генетические факторы могут играть важную роль в определении фенотипического проявления и общего риска развития НАЖБП [2]. В эпидемиологических и генетических исследованиях показана связь морфологической стадии НАЖБП и наследственных факторов. В настоящее время выделяют три гена, ассоциированных с НАЖБП: PNPLA3, TM6SF2 и GCKR. Вместе с генами, отвечающими за инсулинорезистентность, депонирование липидов, воспаление и фиброгенез в гепатоцитах, они определяют фенотип жировой болезни печени. Участие генетических факторов в развитии НАЖБП было доказано у лиц с инсулинорезистентностью и избыточной массой тела.
Кластеры НАЖБП в семьях с определенными генетическими вариациями генов TM6SF2, PNPLA3, NCAN и PPP1R3B или изменениями рядом с ними увеличивают наследуемость НАЖБП до 27%. Одним из генетических вариантов, связанных с НАЖБП, является миссенс-мутация в гене 3, содержащем палатиноподобный фосфолипазный домен (PNPLA3). PNPLA3 оказывает сильное влияние не только в отношении накопления жира в печени, но также и на склонность к развитию более тяжелого гистологического поражения печени, независимо от степени ожирения или наличия диабета [2].
Установлена возможность быстрого прогрессирования НАЖБП при наличии аллеля адипонутрина rs738409 и отсутствии ожирения, сахарного диабета, дислипидемии и других компонентов метаболического синдрома. Изменения в генах, регулирующих процессинг и синтез липопротеинов очень низкой плотности, были сопряжены с частотой выявления НАЖБП [2].
Белок-переносчик эфиров холестерина играет ключевую роль в обратном транспорте холестерина, посредством которого последний проникает из периферических тканей обратно в печень – место его синтеза. L. Adams и соавт. (2012) описали два однонуклеотидных полиморфизма кодирующего гена – rs12447924 и rs12597002, ассоциированные с повышенным риском стеатоза печени; при этом степень риска у лиц с НАЖБП без ожирения была выше [10].
Стеролрегулирующий элементсвязывающий фактор‑2 (SREBF‑2) – ген SREBF‑2 кодирует стеролрегулирующий элементсвязывающий протеин, контролирующий его клеточный синтез, захват и экскрецию: повышение продукции SREBF‑2 в печени коррелирует с тяжестью НАЖБП [2].
Эпигенетические модификации определяются как наследственное, но обратимое явление, влияющее на экспрессию генов без изменения последовательности ДНК. Они включают метилирование ДНК, модификации гистонов и микро-РНК, а также могут играть роль в патогенезе НАЖБП. Эпигенетические модификации, как полагают, являются ключевым детерминантом НАЖБП, и их изучение предоставляет новые возможности для управления заболеваемостью и смертностью от гепатоцеллюлярной карциномы, хотя имеющиеся данные весьма скудны. Необходимы всесторонние исследования относительно применения эпигенетических факторов с целью диагностики и терапии НАЖБП [2].
Согласно данным исследования GWAS Xu et al. (2019), во время прогрессирования НАЖБП активность генов, связанных с биосинтезом липидов, клеточным апоптозом и воспалением, увеличивалась, тогда как генов, связанных с реакцией повреждения ДНК, биосинтезом холестерина и углеводным обменом, снижалась. В других исследованиях была показана корреляция между статусом метилирования или ацетилирования различных гистонов и митохондриальных белков и воспалительной активностью при НАСГ. Кроме того, печеночная экспрессия различных микро-РНК, таких как miR‑122, miR‑335, miR‑29c, miR‑34a, miR‑155 и miR‑200b, участвует в патогенезе НАЖБП, их можно использовать в качестве биомаркеров заболевания [27].
По данным NHANES III, общая тенденция к увеличению распространенности заболевания наблюдается с возрастом. Так, распространенность НАЖБП у мужчин в возрасте от 30 до 40 лет составляет 16,1%, от 41 до 50 лет – 22,3%, старше 60 лет – 27,6%; при этом максимальная распространенность заболевания отмечается в возрасте от 51 до 60 лет – 29,3% [13].
Связь ожирения и НАЖБП обусловлена следующим. Жировая ткань является одним из основных чувствительных к инсулину органов, и процесс дифференциации преадипоцитов в адипоциты индуцируется инсулином [25]. Внутри жировой ткани инсулин стимулирует синтез триглицеридов и ингибирует липолиз, усиливая активность липопротеинлипазы. Она является наиболее чувствительным путем действия инсулина, облегчая поглощение свободных жирных кислот и транспорт глюкозы, ингибируя гормоночувствительную липазу и увеличивая экспрессию генов липогенных ферментов.
Ожирение и нарушение действия инсулина в адипоцитах приводят к неспособности подавления липолиза, стрессу адипоцитов, рекрутингу и инфильтрации макрофагами жировой ткани с последующим высвобождением провоспалительных адипоцитокинов, главным образом фактора некроза опухоли (TNF), интерлейкина (IL)‑6, моноцитарного хемоаттрактантного белка‑1, резистина и ингибитора активатора плазминогена‑1. Эти адипоцитокины способствуют нарушению передачи сигналов инсулина через ядерный фактор-κB (NF-κB) и c-Jun N-терминальную киназу (JNK), созданию порочного круга в жировой ткани и формированию инсулинорезистентности (ИР) в других чувствительных к инсулину тканях, в то время как уровень защитных адипокинов (снижающих ИР), таких как адипонектин, у пациентов с НАЖБП снижается. Уровень адипонектина в сыворотке крови при НАЖБП обычно снижается, тогда как уровень лептина повышается, что способствует возникновению профибротической среды при фиброзе.
Цитокиновые сигналы макрофагов, активные формы кислорода (АФК) и изменение соотношения адипонектин/лептин в пользу последнего ведут к активации звездчатых клеток печени и гиперпродукции экстрацеллюлярного матрикса, т.е. к фиброгенезу при НАЖБП [1].
В прогрессировании НАЖБП и развитии фиброза печени принимают участие разные факторы роста, стимулирующие процессы фиброгенеза путем усиления образования коллагена и соединительной ткани в органе: трансформирующий фактор роста-b (TGF-b), инсулиноподобный фактор роста, тромбоцитарный фактор роста (PDGF).
В последние годы в нескольких когортных исследованиях продемонстрирована связь между изменением массы тела и заболеваемостью НАЖБП. Было показано, что даже незначительное увеличение массы тела на 2 кг в пределах нормы повышает риск развития НАЖБП. Общая распространенность ожирения при НАЖБП в мире составляет 51% [2].
Следует отметить, что существует тип «худой» НАЖБП, который отмечается у пациентов без ожирения. Механизм нежировой неалкогольной болезни печени является сложным, связан с генетическими факторами и обусловлен диетой с высоким содержанием жиров и фруктозы. Пациенты с «мышечной» НАЖБП всегда молодого возраста, часто ведут малоподвижный образ жизни и имеют сниженную чувствительность к инсулину, высокий уровень триглицеридов и высокий риск развития сердечно-сосудистых заболеваний. Тем не менее механизмы прогрессирования стеатоза в фиброз при «худой» неалкогольной болезни печени и НАЖБП одинаковы [3].
Калорийность рациона и специфическое потребление питательных веществ имеют решающее значение для развития НАЖБП, в частности стеатоза печени. Диета, содержащая высокий уровень холестерина, большое количество фруктозы (диета быстрого питания, или фаст-фуд) и насыщенных жиров вызывает ожирение, ИР, развитие стеатоза печени с минимальным воспалением без фиброза. У подростков большее потребление фруктозы связано с множественными маркерами кардиометаболического риска, но, по-видимому, эти отношения опосредованы висцеральным ожирением [10]. Диета с высоким содержанием жиров приводит к подавлению воспалительных процессов в кишечнике и усилению этого механизма в печени.
Cреди диетических факторов фруктоза имеет первостепенное значение, поскольку она является одновременно субстратом и мощным индуктором печеночного синтеза жирных кислот и триглицеридов (липогенез de novo – DNL) из-за активации ключевых факторов транскрипции, таких как SREBP‑1c и ChREBP. Предполагается, что сахара стимулируют DNL и запускают воспалительный ответ, приводящий к апоптозу гепатоцитов через c-Jun-N-терминальный путь. Следует отметить, что гепатоциты подвергаются воздействию более высоких концентраций фруктозы, чем клетки других тканей, поскольку после абсорбции фруктоза напрямую поступает в печень через воротную вену.
Фруктоза, которая истощает внутриклеточный аденозинтрифосфат, при отсутствии инсулина трансформируется в липид, увеличивая таким образом жировые отложения, и вносит свой вклад в развитие НАЖБП и НАСГ. Об этом также свидетельствует сильная связь сахарного диабета 2 типа и НАСГ у лиц, потребляющих много фруктозы, содержащейся в безалкогольных напитках. Истощение печеночного аденозинтрифосфата обусловливает митохондриальную дисфункцию, образование АФК и развитие воспаления, а также усиливает стресс эндоплазматического ретикулума с последующей активацией связанной со стрессом JNK, способствующей апоптозу гепатоцитов, что является отличительным признаком НАСГ.
Фруктоза и определенные специфические рецепторы (например, связанный с G-белком рецептор хемокинов – CX3CR1) могут регулировать кишечную проницаемость при НАЖБП, приводя к прогрессированию заболевания. Фруктоза безалкогольных напитков способствует развитию жировой дистрофии печени и повышению уровня С-реактивного белка [12].
Реализация ИР осуществляется, прежде всего, на уровне гепатоцитов. При нарушении функционального состояния гепатоцитов, развитии воспаления значительно снижается активность многих белков-транспортеров, в первую очередь происходят нарушения в мембранных белках – транспортерах глюкозы. Это приводит к формированию ИР, провоцирующей развитие системной сосудистой воспалительной реакции.
Липидный гомеостаз печени жестко контролируется сложной системой сигнальных/транскрипционных путей, регулируемых гормонами, факторами транскрипции и ядерными рецепторами, при этом передача сигналов инсулина играет ключевую роль. Нарушение передачи сигналов инсулина на уровне жировой ткани и печени способствует тому, что у пациентов с ожирением или сахарным диабетом 2 типа развитие ИР приводит к неингибированному липолизу в жировой ткани. Это вызывает чрезмерный приток неэтерифицированных жирных кислот в печень, где они поглощаются гепатоцитами при помощи транспорта жирных кислот через белок 2 (FATP2), FATP5 и другие транспортные белки, такие как белок, связывающий жирные кислоты (FA), и кавеолин‑1.
Роль железа в прогрессировании НАСГ остается спорной, но есть предположения, что железо может принимать определенное участие в патогенезе НАСГ. В мышиной модели db/db (особи с дефицитом рецептора лептина и генетическим ожирением) перегрузка железом обусловливала окислительный стресс в печени, активацию иммунных клеток и повреждение везикул гепатоцитов, что приводило к НАСГ. Сообщалось, что у пациентов с НАСГ повышена абсорбция железа в двенадцатиперстной кишке за счет активации транспортера двухвалентного металла 1, и что отложение железа в гепатоцитах при НАСГ способствует прогрессированию заболевания за счет увеличения выработки АФК и более высокой частоты апоптоза гепатоцитов. Отложение железа в гепатоцитах также ассоциировано с повышенным риском фиброза печени [13].
Недавно несколько исследовательских групп выявили, что липотоксичность в гепатоцитах связана с усилением воспалительного ответа из-за высвобождения внеклеточных пузырьков – экстрацеллюлярных везикул, происходящих из клеток (EVs). Эти EVs содержат несколько биоактивных молекул (включая белки, липиды и нуклеиновые кислоты), которые служат межклеточными мессенджерами и могут модулировать прогрессирование резидентных мононуклеарных клеток, активирующих НАСГ. Экстрацеллюлярные везикулы также могут передавать сигналы другим клеткам: эндотелиальным клеткам, активирующим ангиогенез, и звездчатым, индуцирующим профиброгенный фенотип. Высвобождение EVs из гепатоцитов представляет собой сложный процесс, вызванный токсической перегрузкой липидами и опосредованный сигнальным каскадом TNF-подобного апоптоз-индуцирующего лиганда (TRAIL) рецептора 2 (TRAIL-R2) и Rho-ассоциированной протеинкиназой 1. EVs были предложены в качестве биомаркеров НАСГ, но необходимы дальнейшие исследования для определения того, можно ли путем выявления EVs в сыворотке крови отличать НАСГ от простого стеатоза [13].
Нарушение сна является независимым фактором риска развития НАЖБП и связано с воспалительными цитокинами (IL‑6 и TNF). Эти цитокины активируются при нарушениях сна и принимают участие в патогенезе НАЖБП за счет повышения липолиза адипоцитов, вызвающего избыточное накопление в печени свободных жирных кислот. Нарушение сна может влиять на ось надпочечники – гипофиз, что, в свою очередь, сказывается на метаболизме кортизола, приводящем к накоплению жира в печени.
Определенное значение в патогенезе НАЖБП имеет снижение синтеза и нарушение транспорта желчных кислот, связанное с повреждением мембран гепатоцитов, ингибированием ферментных систем и воспалением.
В настоящее время активно изучается так называемая энтерогепатическая ось патогенеза НАЖБП. На уровне энтероцитов также происходят метаболические нарушения, связанные с активностью так называемых toll-подобных рецепторов (TLR) 3 типа, CD28. TLR3 – мембранный белок, относится к группе toll-подобных рецепторов, обеспечивающих функционирование врожденного иммунитета. TLR3 играет важную роль в реализации хронического системного воспаления и патогенезе НАЖБП.
По данным проведенных исследований, у пациентов с НАЖБП выявляют более высокие титры антител (IgG) к ряду кишечных бактерий, в частности, Escherichia coli HA116, Bacteroides fragilis, Bifidobacterium thermophilium и Klebsiella oxytoca.
У пациентов с НАЖБП чаще, чем у здоровых лиц, отмечается повышенная проницаемость кишечной стенки, при этом риск ее формирования увеличивается в 30 раз и более на стадии НАСГ. Проницаемость кишечного эпителиального барьера ассоциирована с уровнем липополисахаридсвязывающего белка, гиперпродукция которого характерна для пациентов с НАЖБП [1].
У людей с ожирением чаще развивается синдром избыточного бактериального роста в кишечнике. Клеточные стенки грамотрицательных бактерий содержат липополисахарид (ЛПС) или эндотоксин, который может активировать каскад воспаления через 4-зависимые и TLR4-независимые пути toll-подобного рецептора. Это приводит к повышенной активности генов для некоторых цитокинов – TNF и IL‑6, индуцибельной синтазы оксида азота, NF-κB, ингибитора NF-κB. Данные медиаторы воспаления вызывают ИР. У лиц с НАЖБП повышены кишечная проницаемость и уровень сывороточного эндотоксина, что приводит к бактериальной транслокации и эндотоксемии. В нормальных условиях эта эндотоксемия быстро «очищается» ретикуло-эндотелиальной системой печени. Однако на фоне патологии печени или при длительном воздействии ЛПС, которые являются гепатотоксинами, в органе запускается каскад морфологических и функциональных изменений, вызывая острый воспалительный ответ и увеличение количества полиморфно-ядерных клеток, выделяющих реактивные кислородные метаболиты, протеазы и другие ферменты, с прогрессирующим повреждением печени и формированием НАСГ.
В печени имеется большое количество клеток врожденной иммунной системы. Эти клетки могут распознавать экзогенные молекулы, которые несут специфические патогенассоциированные молекулярные паттерны, и эндогенные вещества с ассоциированными с опасностью молекулярными паттернами через их рецепторы распознавания паттернов, в том числе TLR и нуклеотидсвязывающие белковые рецепторы, содержащие домен олигомеризации. Взаимодействие этих рецепторов с бактериальными продуктами приводит к активации нескольких путей воспаления, включая воспалительные процессы, которые, в свою очередь, активируют каспазу‑1, расщепляющую про-IL‑1β и про-IL‑18 в провоспалительные цитокины.
Повышенное поступление эндотоксинов кишечных бактерий в портальную систему вызывает активацию клеток Купфера, которые индуцируют выработку TGF-β и последующую активацию звездчатых клеток, приводя к формированию фиброза печени. Кроме того, будучи основными прекурсорами миофибробластов, звездчатые клетки также являются преобладающими мишенями, через которые TLR4-лиганды способствуют фиброгенезу. Звездчатые клетки могут также играть важную роль в создании воспалительного каскада в печени, связанного с эндотоксемией. TLR4 активирует не только клетки Купфера, но и звездчатые клетки; в то время как клетки Купфера участвуют в фиброгенезе посредством продуцирования провоспалительных и профиброгенных медиаторов, звездчатые клетки, как указано, являются основным источником внеклеточной матрицы в фиброзном процессе. Активированные звездчатые клетки очень чувствительны к ЛПС через TLR4-зависимый путь.
Под влиянием генетических (полиморфизмы адипонутрина, белка – переносчика эфиров холестерина, SREBF‑2) и пищевых факторов (холестерин, фруктоза), а также нарушений передачи инсулинового сигнала формируется ключевое патогенетическое звено НАЖБП – ИР. Последняя способствует как липолизу, так и липогенезу, обусловливающим перегрузку печени свободными жирными кислотами. Последствиями этой перегрузки являются инициация дополнительной «печеночной» ИР, стресс эндоплазматического ретикулума, активация NF-κB, жировая дистрофия гепатоцитов и окисление свободных жирных кислот, ведущее к гиперпродукции АФК. Стресс эндоплазматического ретикулума усиливает синтез триглицеридов, способствует апоптозу гепатоцитов и активирует JNK и макрофаги. NF-κB стимулирует продукцию TNF, блокирующего инсулиновые рецепторы и нарушающего продукцию адипонектина. Цитокиновые сигналы макрофагов, АФК и изменение соотношения адипонектин/лептин в пользу последнего ведут к активации звездчатых клеток печени и гиперпродукции экстрацеллюлярного матрикса, т.е. фиброгенезу (рис. 4).
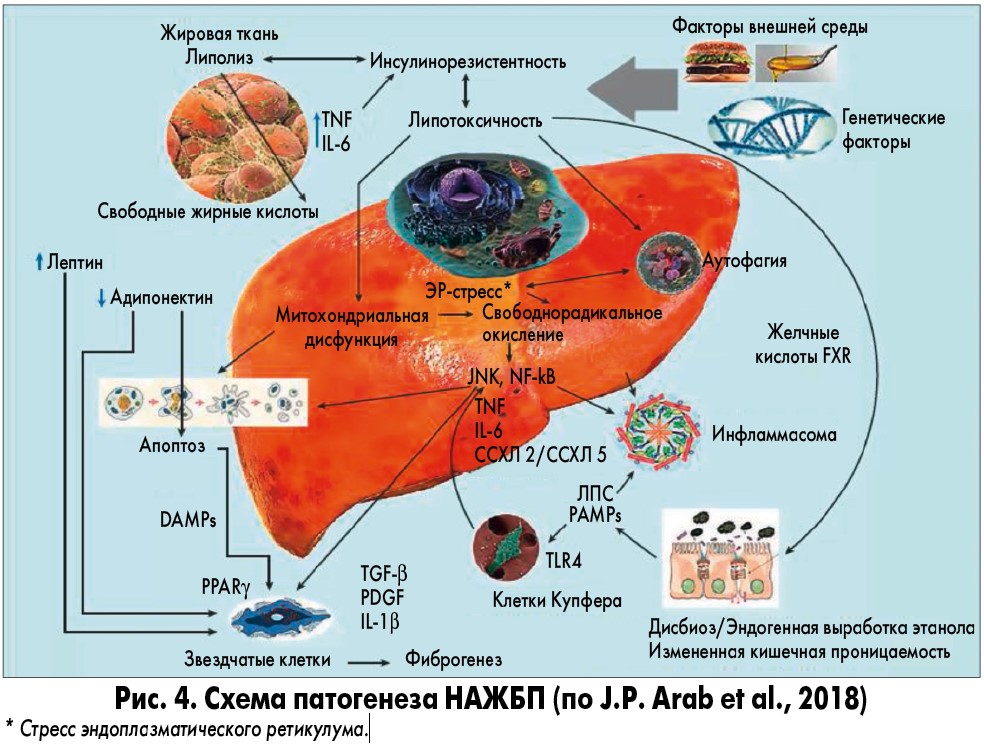
В настоящее время, несмотря на прогресс в понимании патогенеза, клинического течения и прогноза НАЖБП, существуют сложности в выборе оптимального препарата для лечения пациентов с данным заболеванием. Причина, в первую очередь, заключается в том, что это мультифакторное заболевание, а пациент с НАЖБП – это пациент с коморбидной патологией. Этиология НАЖБП сложна, связана с множеством факторов, а клинические проявления и особенности прогрессирования на разных стадиях заболевания и у разных пациентов не совпадают, что увеличивает сложность лечения.
Важной составляющей патогенетического лечения при НАЖБП является предупреждение прогрессирования заболевания от простого стеатоза до фиброза, поэтому предпочтение отдается препаратам природного происхождения с выраженными антифибротическими свойствами. Гепаризин® – единственный гепатопротектор с доказанным прямым антифибротическим и антиканцерогенным действием, который позиционируется как средство для патогенетической терапии при НАЖБП.
Гепаризин® имеет две формы выпуска: ампулы для внутривенного введения и капсулы для перорального применения. Недавно компания «Валартин Фарма» представила Гепаризин® в капсулах с повышенной в 2 раза концентрацией ингредиентов – Гепаризин форте. Он содержит 50 мг глицирризина, 50 мг глицина и 50 мг метионина. Такая дозировка способствует повышению пациентами комплайенса, так как суточная доза будет достигнута при приеме 3 капсул в день (против 6 капсул в день при применении препарата Гепаризин®). Гепаризин форте принимают по 1 капсуле 3 раза в день, длительность курса подбирают индивидуально, минимальный курс – 1 мес.
Прямое антифибротическое действие глицирризина основано на снижении концентрации TNF и TGF-β1 (цитокинов, приводящих к индукции фиброгенеза), подавлении активации звездчатых клеток, угнетении избыточного синтеза коллагена I и III типа, активации коллагеназы (ММРs), что приводит к дегрануляции избыточных отложений коллагена в экстрацеллюлярном матриксе, ингибированию каспазы‑3, -8, снижению процессов апоптоза гепатоцитов [6]. Супрессивное воздействие препарата Гепаризин® на TNF и каспазу‑3 обусловливает его противовоспалительный и гепатопротекторный эффект.
Антивирусное действие глицирризина заключается в ингибировании вирусной репликации и регуляции иммунных реакций. Механизмом действия глицирризина при лечении вирусного гепатита С является дозозависимое ингибирование первичных частиц вируса, а также подавление экспрессии и функции основного ядерного белка, который оказывает провоспалительное и цитопатическое действие на гепатоциты.
Глицирризин способен усиливать некоторые иммунные функции, такие как продукция интерферонов и активность натуральных киллеров, а также модулировать реакции роста лимфоцитов путем повышения выработки IL‑2 (активация врожденного иммунитета).
Аминокислота глицин, входящая в состав препарата Гепаризин®, способствует снижению окислительного стресса, уменьшает воспаление и повреждение гепатоцитов свободными радикалами за счет увеличения синтеза глутатиона, нейтрализует токсическое воздействие этанола на организм, участвует в детоксикации аммиака, хелатообразовании, оказывает антиастеническое, антифибротическое, иммуномодулирующее, антиапоптотическое, энергосберегающее, противовирусное действие, препятствует возникновению побочных эффектов при длительном применении глицирризина.
Цистеин/метионин – серосодержащие аминокислоты, выступающие в роли антиоксиданта и детоксиканта (SH-группы инактивируют свободные радикалы), являются предшественниками глутатиона и метионина, оказывают антигипоксическое, антиоксидантное, детоксикационное, липотропное действие [6, 7]. Предотвращают возникновение побочных эффектов при длительном применении глицирризина.
Глицирризин (главный компонент препарата Гепаризин®) представляет собой соединение калиевой и кальциевой соли глицирризиновой кислоты, которое является тритерпеновым гликозидом, образованным одной молекулой глицирретовой кислоты и двумя молекулами глюкуроновой кислоты. Наличие агликана (глицирретовой кислоты), по своей структуре сходного с глюкокортикоидами, обусловливает гормоноподобное действие глицирризина.
Противовоспалительное действие препарата связано с псевдокортикостероидным (гормоноподобным) эффектом: глицирризин ингибирует 11-β-оксистероиддегидрогеназу, которая превращает глюкокортикоид кортизол в его неактивную форму кортизон, что приводит к увеличению концентрации эндогенного кортизола в крови и одновременной активации минералокортикоидных рецепторов.
Гепаризин® является синергистом синтетических глюкокортикоидных препаратов за счет блокирования расщепляющих их ферментов, угнетает провоспалительные цитокины: TNF, IL‑6, NF-κB в макрофагах, стимулирует синтез противовоспалительных цитокинов (IL‑2, -10, -12), селективно блокирует фосфолипазу А – метаболический фермент синтеза арахидоновой кислоты, в связи с чем блокируется образование простагландинов, лейкотриенов и других факторов воспаления.
Гепаризин® может ослаблять воспалительные реакции и модулировать врожденный иммунный ответ. В зависимости от исходного состояния иммунной системы реализуются два пути действия препарата – либо иммуностимуляция, либо иммуносупрессия. Иммунорегулирующее действие препарата Гепаризин® обусловлено регуляцией активации Т-лимфоцитов, индукцией интерферона-γ, активацией натуральных киллеров, дифференциацией Т-лимфоцитов тимуса.
Tingting Yan и соавт. (2018) показали, что глицирризин посредством восстановления гомеостаза желчных кислот и ингибирования воспалительного повреждения может быть терапевтической опцией для лечения пациентов с НАСГ. По данным этих авторов, глицирризин значительно ослабил экспрессию мРНК фиброгенетического гена, включая TGF-β1, тканевые ингибиторы металлопротеиназ 1 и 2, коллагена 1 и 2 и матриксных металлопротеиназ 2 и 9. Эти результаты свидетельствуют, что введение глицирризина способствует значительному уменьшению фиброгенеза печени, связанного с НАСГ [23].
Европейская исследовательская группа SNMC (M.P. Manns, H. Wedemeyer, A. Singer et al., 2012) cообщила о проведении III фазы исследования, направленного на подтверждение эффективности и безопасности применения глицирризина у пациентов, которым раньше не проводили терапию на основе интерферона + рибавирина, с оценкой биохимических и гистологических эффектов через 52 недели. Исследование было проведено в 73 центрах 11 европейских стран. После рандомизированного двойного слепого плацебо-контролируемого сравнения глицирризина, вводимого внутривенно 5 или 3 раза в неделю, и плацебо 5 раз в неделю в течение 12 недель 379 пациентам, последовало рандомизированное открытое сравнение. Глицирризин применяли внутривенно 5 и 3 раза в неделю в течение 40 недель. Первичными конечными точками были: доля пациентов со снижением уровня аланинаминотрансферазы ≥50% после 12 недель двойной слепой фазы и доля пациентов с уменьшением некровоспаления через 52 недели по сравнению с исходным уровнем. Доля пациентов с уменьшением некровоспаления через 52 недели при введении глицирризина 5 раз в неделю составила 44,9%, 3 раза в неделю – 46,0% соответственно. Глицирризин обеспечивал значительно более высокое снижение уровня аланинаминотрансферазы по сравнению с плацебо через 12 недель терапии, уменьшение некровоспаления и фиброза после 52-недельного лечения. Авторы отмечали хорошую переносимость глицирризина [18].
Результаты многочисленных рандомизированных контролируемых исследований свидетельствуют о том, что препарат SNMС (глицирризин + глицин + цистеин) значительно улучшает функцию печени у пациентов с циррозом печени: 252 пациента с циррозом печени получали SNMС по 2 капсулы 3-4 раза в день в течение 3 месяцев. У 80 (32%) пациентов выявлена нормализация биохимических показателей, у 167 (66%) – улучшение показателей в 2 раза и более, у 5 (2%) эффект не достигнут. Общая эффективность терапии составила 98% (Yue Sihong, 1998) [26].
Гепаризин® является полным аналогом японского препарата SNMC. Гепаризин® способствует уменьшению выраженности стеатоза, липотоксичности и фиброза печени; оказывает метаболический эффект, обеспечивая нормализацию липидного спектра, снижает ИР, что дает основание считать его патогенетическим средством с доказанным антифибротическими свойствами для лечения пациентов с НАЖБП.
Собственные наблюдения у больных с алкогольным фиброзом печени и НАЖБП также подтверждают антифибротическое, противовоспалительное действие препарата Гепаризин® [6-8].
Cписок литературы находится в редакции.
Тематичний номер «Гастроентерологія. Гепатологія. Колопроктологія» № 2 (52), травень 2019 р.