


31 травня, 2019
Сучасний погляд на курацію пацієнтів зі змішаними аутоімунними хворобами печінки
 Аутоімунний печінковий перехресний синдром (ПС), або оверлап-синдром характеризується проявом в одного і того ж хворого ознак двох різних аутоімунних захворювань печінки, які, найімовірніше, мають спільний генез [5, 17]. Аутоімунний гепатит (АІГ), як найчастіша складова змішаних аутоімунних хвороб печінки, має два основних варіанти фенотипу, у яких прояви класичного захворювання змішуються з симптомами первинного біліарного холангіту (ПБХ) або первинного склерозуючого холангіту (ПСХ). Значно рідшим є варіант поєднання ПБХ і ПСХ [3, 16].
Аутоімунний печінковий перехресний синдром (ПС), або оверлап-синдром характеризується проявом в одного і того ж хворого ознак двох різних аутоімунних захворювань печінки, які, найімовірніше, мають спільний генез [5, 17]. Аутоімунний гепатит (АІГ), як найчастіша складова змішаних аутоімунних хвороб печінки, має два основних варіанти фенотипу, у яких прояви класичного захворювання змішуються з симптомами первинного біліарного холангіту (ПБХ) або первинного склерозуючого холангіту (ПСХ). Значно рідшим є варіант поєднання ПБХ і ПСХ [3, 16].
Етіологія хронічних захворювань печінки, перебіг яких характеризується наявністю ПС, до теперішнього часу не вивчена. Розглядається фактор спадкової схильності, а також ціла низка тригерних механізмів, здатних запустити кожне з двох захворювань, які включені в ПС. Патогенні механізми, що відповідають за поєднаний перебіг аутоімунних захворювань печінки, до кінця не зрозумілі. На жаль, на сучасному етапі ми не маємо стратегій лікування, ефективність яких є доведеною. Останній факт, передусім, пов’язаний із невеликою поширеністю цих захворювань і ще більш рідкісним їх поєднанням [6, 12]. Частота перехресту АІГ із ПБХ становить 1-14,2%, а частота перехресту з АІГ з ПСХ – 1,4-17% [4, 8, 10]. Згідно з даними останніх досліджень вважається, що поширеність істинного оверлап-синдрому з наявністю ПБХ, який чітко відповідає сучасним критеріям, становить не більше 2% [8]. Визначення ПС необхідне переважно для ідентифікації пацієнтів, які погано реагують на стандартне лікування.
Найчастішим варіантом ПС є поєднання АІГ і ПБХ, що може спостерігатися у двох варіантах: одночасному та послідовному [16, 17]. При одночасному ознаки обох захворювань виникають майже в один той самий час. При другому варіанті є 2 різновиди. Один із них характеризується початком захворювання з ознаками АІГ, після чого з’являються ознаки ПБХ із серологічними маркерами (антимітохондріальні антитіла до піруватдегідрогеназного комплексу – АМА-М2) і біохімічними змінами у вигляді зростання рівня ферментів холестазу. За іншого варіанту послідовного ПС спочатку у пацієнтів виникає клінічна картина ПБХ, після чого з’являються ознаки АІГ (високий цитоліз, позитивні антинуклеарні антитіла, антигладеньком’язові антитіла тощо) [5, 16]. У двох дослідженнях показано, що близько у 2,5% хворих на ПБХ розвивається гострий АІГ, але за даними найбільшого дослідження встановлено, що тільки у 8 з 1476 пацієнтів з ПБХ пізніше розвинувся АІГ [6, 8].
Враховуючи специфічні особливості ведення таких хворих, важливість правильного та своєчасного визначення наявності оверлап-синдрому складно переоцінити. Тому треба знати та використовувати стандартні міжнародні критерії. Найбільш часто застосовуваною та рекомендованою всіма останніми міжнародними настановами є система під назвою «Паризькі критерії» [5, 7, 16]. Відповідно до цієї системи діагноз ПС ПБХ/АІГ при початковому розвитку ПБХ вимагає наявності двох із таких трьох діагностичних критеріїв:
рівень аланінамінотрансферази мінімум у 5 разів перевищує верхню межу норми (ВМН);
показник IgG щонайменше в 2 рази вищий за ВМН та/або позитивний тест на антитіла до гладеньком’язових клітин;
наявність ознак помірного або тяжкого інтерфейсного гепатиту* за даними гістологічного дослідження біоптату печінки.
Діагноз ПС ПБХ/АІГ при початковому розвитку АІГ вимагає наявності двох із таких трьох діагностичних критеріїв:
- активність лужної фосфатази ≥2×ВМН або гамма-глутамілтрансферази ≥5×ВМН;
- наявність АМА;
- виражені ураження жовчних проток у біопсії препарату печінки.
Встановлено, що ці критерії з високою чутливістю (92%) і специфічністю (97%) дозволяють виявити пацієнтів із клінічним діагнозом варіанта АІГ-ПБХ [6, 12].
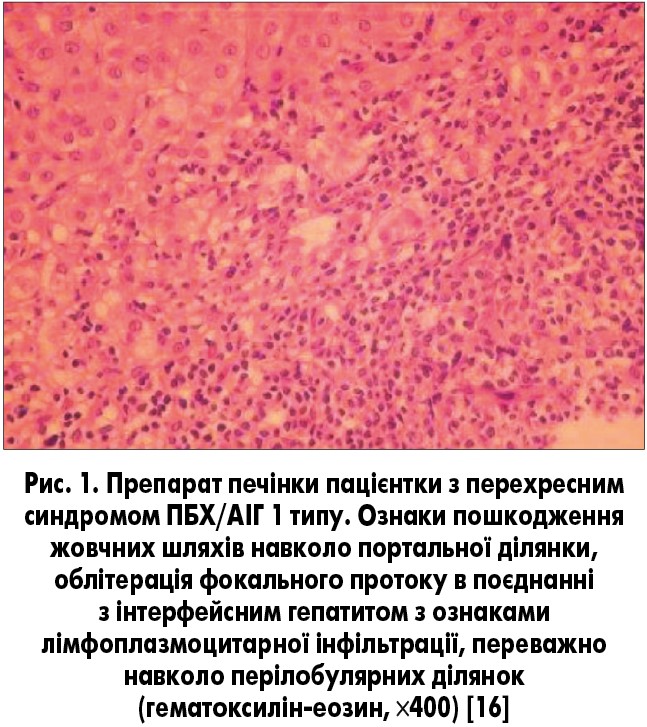 Важливо, що термін «перехресний синдром» не повинен стосуватися пацієнтів з ПБХ, у яких виявляються позитивні лабораторні маркери АІГ, і легким ступенем інтерфейсного гепатиту (рис. 1), оскільки такі ознаки можуть зустрічатися при ПБХ [6, 12].
Важливо, що термін «перехресний синдром» не повинен стосуватися пацієнтів з ПБХ, у яких виявляються позитивні лабораторні маркери АІГ, і легким ступенем інтерфейсного гепатиту (рис. 1), оскільки такі ознаки можуть зустрічатися при ПБХ [6, 12].
За наявності ознак ПС перебіг захворювання є більш тяжким, ніж при наявності однієї хвороби, і характеризується вищою частотою поширеного фіброзу на момент встановлення діагнозу (навіть у пацієнтів молодого віку). Клінічний перебіг ПС (АІГ/ПБХ), як правило, більшою мірою визначається АІГ. Темпи прогресування в цьому випадку вищі, ніж при ізольованому ПБХ, та зумовлені вираженістю запально-некротичних змін у паренхімі печінки [5, 17].
На етапі сформованого цирозу печінки встановити діагноз ПС за гістологічною картиною тканини печінки майже неможливо. У цьому випадку клінічне значення має своєчасна й адекватна корекція ускладнень цирозу печінки [5]. У більшості публікацій відзначається також несприятливий прогноз щодо біохімічної відповіді на прийом урсодезоксихолевої кислоти (УДХК), прогресування фіброзу і смерті, пов’язаної з печінковою недостатністю [6].
Незважаючи на відсутність контрольованих досліджень, у рекомендаціях Європейської асоціації з дослідження печінки (EASL) на підставі результатів невеликих досліджень зазначено про необхідність додавання кортикостероїдів (преднізолону або будесоніду) або на момент встановлення діагнозу ПС, або d разі недостатньої біохімічної відповіді після 3 міс застосування УДХК [6, 7, 16]. Цікавими є результати великого багатоцентрового дослідження E. Ozaslan (2014) за участю 88 пацієнтів, у ході якого 30 хворих як терапію першої лінії отримували УДХК, 58 – комбінацію УДХК та імунодепресантів (преднізон±азатіоприн). У пацієнтів із помірним перипортальним гепатитом ефективність монотерапії УДХК і комбінованої терапії, судячи з біохімічної відповіді, була подібною (80%), тоді як у хворих на гепатит тяжкого ступеня ефективність монотерапії УДХК була значно нижчою (14 vs 71%). Наявність поширеного фіброзу була пов’язана з відсутністю відповіді на комбіновану терапію, але не на монотерапію УДХК. У результаті терапії другої лінії імуносупресивними препаратами (циклоспорином, такролімусом і мікофенолату мофетилом) досягнуто біохімічної ремісії у половини пацієнтів з відсутністю відповіді на початкову імуносупресію [14]. Ці дані свідчать на користь доцільності застосування комбінації УДХК та імунодепресантів як терапії першої лінії у пацієнтів із ПБХ і тяжким перісептальним гепатитом.
Існує думка, що при тривалому лікуванні за наявності відповіді дозу імуносупресивних препаратів можна зменшити, а частота успішного припинення лікування є вищою, ніж при класичному АІГ [6]. У пацієнтів із ПБХ, у яких розвивається АІГ (послідовний варіант) та які отримували УДХК, необхідно застосовувати імунодепресанти [6, 8, 18].
В останніх рекомендаціях щодо терапії ПСХ пропонується розглядати обетіхолеву кислоту як препарат, що використовується при недостатній ефективності УДХК. Слід зазначити, що стосовно ПС ПБХ/АІГ такої рекомендації немає [6, 18].
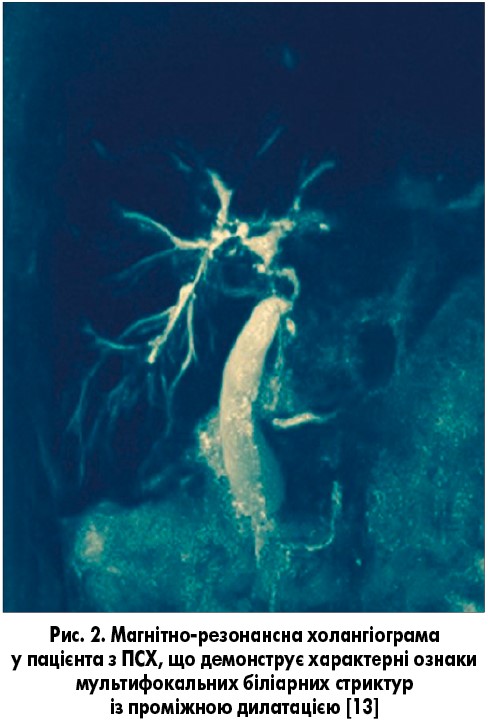 ПС ПСХ/АІГ переважно зустрічається у дітей, підлітків і дорослих молодого віку [1, 9]. Синонімом є термін «аутоімунний склерозуючий холангіт». Його характеристики включають клінічні, біохімічні та гістологічні особливості АІГ і холангіографічні ознаки, характерні для ПСХ (рис. 2) [8, 9]. Ретроспективна діагностика ПС ПСХ/АІГ, згідно з сучасними критеріями, дозволила встановити його наявність у 1,4-17% пацієнтів [13]. У дітей і підлітків з ПС ПСХ/АІГ частіше виявляли хронічні запальні захворювання кишечнику, вони частіше були позитивними за показником p-ANCA у сироватці, ніж ті, хто має тільки АІГ. Трансамінази сироватки зазвичай є вищими при АІГ, але лабораторні показники холестазу можуть бути нормальними при оверлап-синдромі ПСХ/АІГ [1].
ПС ПСХ/АІГ переважно зустрічається у дітей, підлітків і дорослих молодого віку [1, 9]. Синонімом є термін «аутоімунний склерозуючий холангіт». Його характеристики включають клінічні, біохімічні та гістологічні особливості АІГ і холангіографічні ознаки, характерні для ПСХ (рис. 2) [8, 9]. Ретроспективна діагностика ПС ПСХ/АІГ, згідно з сучасними критеріями, дозволила встановити його наявність у 1,4-17% пацієнтів [13]. У дітей і підлітків з ПС ПСХ/АІГ частіше виявляли хронічні запальні захворювання кишечнику, вони частіше були позитивними за показником p-ANCA у сироватці, ніж ті, хто має тільки АІГ. Трансамінази сироватки зазвичай є вищими при АІГ, але лабораторні показники холестазу можуть бути нормальними при оверлап-синдромі ПСХ/АІГ [1].
Таким чином, у пацієнтів з АІГ, які мають ознаки холестазу та/або є стійкими до імуносупресії, слід виключати ПС ПСХ/АІГ.
При оверлап-синдромі ПСХ/АІГ також використовувалася УДХК в поєднанні з імуносупресивними схемами [9]. Таких пацієнтів лікували з УДХК (15-20 мг/кг/добу), преднізолон (0,5-1 мг/кг/добу і 50-75 мг азатіоприну з хорошою біохімічною реакцією.
При проведенні початкового лікування пацієнт із ПС ПСХ/АІГ часто відповідає на іммуносупресивну терапію [1, 13]. Лабораторні показники в більшості випадків нормалізуються протягом кількох місяців лікування, хоча довгостроковий прогноз є гіршим, ніж при ізольованому АІГ, оскільки ураження жовчних протоків продовжує прогресувати приблизно у 50% пацієнтів.
Оптимальна тривалість імуносупресивної терапії не визначена, але її припинення можливе лише при значному зменшенні запального процесу за даними гістологічного дослідження. Відповідь на лікування можна контролювати, оцінюючи концентрацію IgG і титри аутоантитіл, коливання яких корелюють з активністю захворювання [5, 6, 16]. Важливо відмітити, що відсутність сироваткових аутоантитіл не виключає ризик розвитку рецидиву. Встановлено, що повна відміна лікування в перші 2 роки зазвичай супроводжується рецидивами [6, 17]. Таким чином, про повне припинення лікування може йтися тільки після фіксації стабільно нормальних функціональних проб печінки протягом 1-2 років, нормальних рівнів IgG і негативних титрів аутоантитіл при відсутності запальної активності за даними біопсії печінки.
Відносно нещодавно почали виділяти окреме захворювання, яке потребує дуже ретельної диференційної діагностики з ПСХ. Це імуноглобулін G4-асоційований холангіт (IgG4-АХ). На цей час IgG4-АХ розглядається як прояв IgG4-асоційованого склерозуючого захворювання та діагностується на підставі сукупної оцінки характерних клінічних, рентгенографічних, серологічних, гістологічних та імуногістохімічних ознак [2]. При цьому гістопатологічним дослідженням надається першочергове значення. Виявлення трьох основних гістологічних ознак (щільного лімфоплазматичного інфільтрату, багатоярусного фіброзу і облітеруючого флебіту) дозволяє вчасно діагностувати захворювання. А при збільшенні співвідношення IgG4-позитивних плазматичних клітин (ППК) / IgG-ППК >40% діагноз IgG4-асоційованого склерозуючого захворювання не підлягає сумніву [2, 9].
IgG4-АХ являє собою біліарне захворювання невідомої етіології, що має біохімічні та холангіографічні особливості, які не відрізняються від ПСХ, найчастіше супроводжується позабіліарними проявами, реагує на терапію кортикостероїдами, часто асоціюється з аутоімунним панкреатитом і характеризується підвищенням сироваткового IgG4 та інфільтрацією IgG4-ППК у жовчних протоках і тканині печінки [2]. На відміну від ПСХ, IgG4-АХ не пов’язаний із запальними захворюваннями кишечнику. Попередні дані свідчать про те, що імунопатогенез IgG4-АХ відрізняється від інших аутоімунних холестатичних захворювань печінки, таких як ПСХ і ПБХ. Переважно зустрічається у чоловіків, середній вік при встановленні діагнозу – 60 років.
Були запропоновані критерії IgG4-АХ [11]. Для встановлення такого діагнозу пацієнт із біліарною стриктурою, що локалізована у внутрішньопечінкових, проксимально позапечінкових і/або інтрапанкреатичних жовчних протоках, повинен мати:
- нещодавнє панкреатичне/біліарне хірургічне втручання в анамнезі,
- або гістологічні ознаки аутоімунного панкреатиту (AIП) / IgG4-АХ,
- або класичні ознаки AIП і підвищений IgG4,
- або два з наведених критеріїв (підвищення IgG4 у сироватці крові; типові рентгенологічні дані ураження підшлункової залози; інші ознаки ураження органів, включаючи склерозуючий сіалоаденіт, ретроперитонеальний фіброз, абдомінальну лімфаденопатію з інфільтрацією IgG4-ППК; >10 IgG4-ППК у біоптатах жовчних проток), а також адекватну відповідь на 4-тижневий курс лікування кортикостероїдами, що дозволяє видалення стента без рецидиву обструктивного холестазу, щоб досягти зниження печінкових показників <2 ВМН і зменшення IgG4 і CA 19-9 [9].
На противагу молодим пацієнтам із ПС ПСХ/АІГ у більшості хворих на IgG4-АХ захворювання дебютує у старшому віці (60 років) жовтяницею (77%) та клінічними проявами АІП (92%). Стриктури виявляються при магнітно-резонансній холангіопанкреатографії або ендоскопічній ретроградній холангіопанкреатографії ізольовано в інтрапанкреатичній ділянці Вірсунгової протоки у 51% хворих. В інших випадках вони розташовані у жовчовивідних протоках. Рівень сироваткового IgG4 значно вищий, ніж при ПСХ. У разі наявності ПСХ холангіографічні особливості проявляються у вузлуватих стриктурах жовчних проток, що мають вигляд дерева з обрізаними гілками, тоді як пацієнти з IgG4-АХ мають сегментарні стриктури і звуження дистальної третини загальної жовчної протоки. При гістологічному вивченні печінки виявляється облітеруючий фіброзний холангіт тільки у хворих на ПСХ; при IgG4-АХ він спостерігається рідко. На відміну від ПСХ для IgG4-АХ типовою є виражена інфільтрація IgG4-ППК портальних трактів печінки [2, 9].
Холангіографічні дослідження у більшості випадків не виявляють поліпшення при ПСХ після стероїдної терапії, на відміну від IgG4-АХ. Крім того, холангіокарцинома розвивається у 10-30% хворих на ПСХ і не характерна для IgG4-АХ [2, 9].
Показано, що імуносупресивне лікування помітно впливає на запальну активність IgG4-АХ, при цьому тривалої ремісії можна досягти вже через 3 міс лікування. Проте тяжкість захворювання може впливати на довгостроковий ефект. Ретроспективний аналіз показав, що пацієнти зі змінами проксимальних позапечінкових і внутрішньопечінкових жовчних проток мають вищий ризик рецидиву після припинення лікування, ніж пацієнти зі стриктурами дистальної жовчної протоки [159]. Кортикостероїди розглядаються як препарати вибору при цьому захворюванні. Використання азатіоприну в дозі до 2 мг/кг/добу має розглядатися у хворих із проксимальними і внутрішньопечінковими стриктурами в разі недостатнього ефекту після терапії кортикостероїдами та при рецидиві. Лікування тривалістю 3 міс може бути достатнім для деяких пацієнтів, але тривала підтримувальна терапія низькими дозами може знадобитися при наявності високого ризику рецидиву [9, 11].
Трансплантація печінки показана пацієнтам із будь-яким типом ПС, якщо хвороба маніфестує за типом фульмінантної печінкової недостатності, і тим, у кого, незважаючи на лікування, розвивається термінальна стадія захворювання печінки. Після трансплантації рецидив АІГ описується в 12-46% випадків, а рецидив ПС ПСХ/АІГ – близько в 70% і частіше при наявності активного запального захворювання кишечнику. Діагностика рецидиву ґрунтується на зміні біохімічних показників, серопозитивності аутоантитіл, розвитку интерфейсного гепатиту за даними гістології та наявності холангіопатії при ПС ПСХ/АІГ. Рецидив може виникнути навіть через кілька років після трансплантації, а успіх лікування значною мірою залежить від ранньої діагностики [2, 6, 9, 16].
За наявності показань при ПСХ проводиться стентування жовчовивідних шляхів.
Таким чином, аутоімунні оверлап-синдроми є дуже складною проблемою сучасної гепатології. Багато в чому ці стани є недостатньо дослідженими, але вони являють собою клінічну реальність, яку необхідно приймати. Існує декілька напрямів подальшого розвитку терапії змішаних аутоімунних захворювань. По-перше, це продовження досліджень щодо виявлення й уточнення етіопатогенетичних чинників, що дозволить призначати відповідне лікування. По-друге, це розроблення і використання нових імуносупресивних агентів. По-третє, дуже важливим аспектом для будь-яких хронічних захворювань печінки, у тому числі ПС, є уповільнення прогресування фіброзу печінки, тому перспективною є розробка нових схем із використанням антифібротичних агентів. Діагностика та лікування таких станів мають проводитися відповідно до чинних міжнародних рекомендацій у спеціалізованих гепатологічних центрах.
Література
- Костырко Е.В., Шумилов П.В. Современные методы лечения детей с аутоиммунными заболеваниями печени // Педиатрическая фармакология. – 2015. – № 12 (6). – С. 679-685.
- Степанов Ю.М., Гайдар Ю.А. Імуноглобулін G4-асоційовані склерозуючі захворювання органів травлення // Журнал НАМН України. – 2015, т. 21. – № 1. – С. 54-60.
- Boberg K.M., Chapman R.W., Hirschfield G.M. et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011; 54: 374-385.
- Bonder A., Retana A., Winston D.M. et al. Prevalence of primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. Clin Gastroenterol Hepatol 2011; 9: 609-612.
- Czaja A.J. The overlap syndromes of autoimmune hepatitis. Dig Dis Sci. 2013; 58(2): 326-343.
- Chazouilleres O., Dalekos G., Drenth J. EASL Clinical Practice Guidelines: Autoimmune hepatitis J of Hepatol, 2015: 63.
- Chazouilleres O., Wendum D., Serfaty L. et al. Long term outcome and response to therapy of primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. J Hepatol, 2006; 44: 400-406.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. Journal of Hepatology 2009; 51: 237-267.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. Journal of Hepatology 2017; 67: 145-172.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. Journal of Hepatology 2009; 51: 237-257.
- Chapman R., Fevery J., Kalloo A. et al. Diagnosis and Management of Primary Sclerosing Cholangitis // Hepatology 2010; 51(2): 660-678.
- Ghazale A., Chari S.T., Zhang L. et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology 2008; 134: 706-715.
- Lindor K.D., Bowlus C.L., Boyer J. et al. Practice Guidance from the American Association for the Study of Liver Diseases: Primary Biliary Cholangitis Hepatology, 2018.
- Marchioni Beery R.M., Forouhar F., Vaziri H. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: a Review Featuring a Women’s Health Perspective. J. Clin. Transl. Hepatol. 2014; 2, I.4: 266-284.
- Ozaslan E., Efe C., Heurgue-Berlot A. et al. Factors associated with response to therapy and outcome of patients with primary biliary cirrhosis with features of autoimmune hepatitis. Clin Gastroenterol Hepatol 2014; 12: 863-869.
- Pamfil C., Candrea E., Berki E. et al. Primary biliary cirrhosis – autoimmune hepatitis overlap syndrome associated with dermatomyositis, autoimmune thyroiditis and antiphospholipid syndrome J Gastrointestin Liver Dis 2015; 24(1): 101-104.
- Silveira M.G. Overlap syndromes of autoimmune liver disease. J Clin Cell Immunnol. 2013; 4: 4.
- Yokokawa J., Saito H., Kanno Y. et al. Overlap of primary biliary cirrhosis and autoimmune hepatitis: characteristics, therapy, and long term outcomes. J Gastroenterol Hepatol 2010; 25: 376-82.
- Zhang H., Li S., Yang J. et al. A meta-analysis of ursodeoxycholic acid therapy versus combination therapy with corticosteroids for PBC-AIH-overlap syndrome: evidence from 97 monotherapy and 117 combinations. Prz Gastroenterol 2015; 10: 148-55.
*Інтерфейсний гепатит (ступінчасті некрози, або перипортальний гепатит) характеризується проникненням запальних лімфоплазмоцитарних інфільтратів у паренхіму печінки з відділенням («відшнуруванням») і руйнуванням ізольованих гепатоцитів або їх невеликих груп.
Тематичний номер «Гастроентерологія. Гепатологія. Колопроктологія» № 2 (52), травень 2019 р.