


2 жовтня, 2019
Ювенільний дерматоміозит
 Дерматополіміозити – група захворювань сполучної тканини аутоімунного характеру із переважним ураженням м’язів, в яку входить ювенільний дерматоміозит (ЮДМ). Це важке захворювання, яке починається у дітей до 16 років, з переважним системним ураженням м’язів, шкірних покривів, порушенням мікроциркуляції та органною патологією із хвилеподібним прогресуючим перебігом, розвитком васкулітів, розповсюдженого кальцинозу та сухожильно-м’язових контрактур.
Дерматополіміозити – група захворювань сполучної тканини аутоімунного характеру із переважним ураженням м’язів, в яку входить ювенільний дерматоміозит (ЮДМ). Це важке захворювання, яке починається у дітей до 16 років, з переважним системним ураженням м’язів, шкірних покривів, порушенням мікроциркуляції та органною патологією із хвилеподібним прогресуючим перебігом, розвитком васкулітів, розповсюдженого кальцинозу та сухожильно-м’язових контрактур.
ЮДМ відрізняється від дерматоміозиту дорослих наявністю поширеного васкуліту, вираженими міалгіями, ураженням внутрішніх органів і високою частотою розвитку кальцинозу.
Розповсюдженість ЮДМ оцінюється близько 4:100 тис, дівчата хворіють частіше, ніж хлопчики, у більшості випадків дебют захворювання спостерігається у віці 4-10 років.
Етіологія ЮДМ невідома, розвиток захворювання вважається багатофакторним, мається на увазі комбінація генетичних факторів та умов середовища. Вагомою причиною є інфекційний агент, тому що розвитку клінічної картини цього захворювання завжди передують вірусні інфекції, що викликані вірусами Коксакі В, парвовірусами, вірусами грипу, парагрипу. Серед бактеріальних збудників виділяють β-гемолітичний стрептокок групи А. Розвиток ЮДМ асоціюється із носійством генів HLA-B8 та DR‑3, а можливо, і HLA-В14 та В40, що говорить про участь генетичних факторів. Це сприяє продукції певних міозитспецифічних антитіл. Частота аутоімунних хвороб у сім’ях дітей із ЮДМ підвищена, при зборі анамнезу виявляються захворювання аутоімунної природи серед близьких родичів.
Патогенетичним фактором ЮДМ є аутоімунний механізм, коли продукуються аутоантитіла проти цитоплазматичних білків та рибонуклеїнових кислот, які входять до складу м’язової тканини і формують мікроангіопатію з залученням капілярів ендомізію. Типовим для ЮДМ є відкладання мембранолітичних комплексів (комплекс компліменту С5b‑9) на стінках внутрішньом’язових капілярів, що може призвести до їх руйнування, м’язової ішемії, перивасцикулярної атрофії та ендомізіального фіброзу. Патогенетичну роль у цьому процесі відіграють також антиендотеліальні антитіла.
Гістологічні зміни спостерігаються переважно у м’язах: виявляється гіперплазія ендотелію судин малого калібру, перивасцикулярна атрофія, відсутність поперекової накресленості м’язів, їх набряк та вакуолізація, явища дегенерації, вогнища мононуклеарної клітинної інфільтрації, ознаки фагоцитозу некротизованих м’язів, інтерстиціальний фіброз. Гістологічні зміни шкіри неспецифічні, спостерігається помірна атрофія епідермісу, явища васкуліту.
Класифікація ЮДМ. Загальноприйнятої класифікації ЮДМ не існує. У дітей переважно спостерігається ідіопатичний (первинний) ЮДМ. Він відрізняється двома особливостями: розвитком васкулітів у початковому періоді, кальцинозом тканин у подальшому, схильністю до гіперергічних реакцій, змінами ферментного та білкового обміну.
У практичній діяльності використовують класифікацію, в основі якої лежать критерії, запропоновані Л.О. Ісаєвою та М.О. Жванією в 1978 р. (таб. 1).
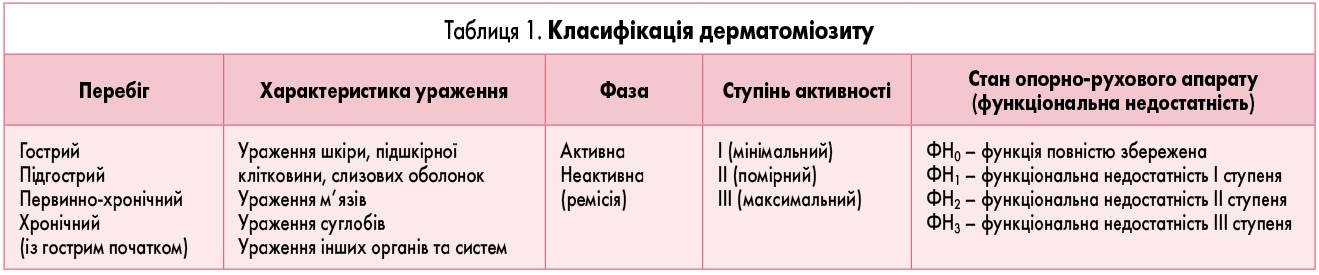
У процесі перебігу захворювання виділяють наступні періоди:
- продромальний – від декількох днів до місяця;
- маніфестний із шкірними, м’язовими та загальними синдромами;
- дистрофічний або кахектичний, термінальний період ускладнень.
Гострий перебіг ЮДМ характеризується значним підвищенням температури тіла, вираженими шкірними проявами, швидко прогресуючою м’язовою слабкістю майже до повної нерухомості, різким больовим та набряковим синдромом, дисфагією, вісцеральними проявами із ураженнями життєво важливих органів, високою запальною активністю крові. Триває цей процес 3-6 тижнів.
Підгострий перебіг захворювання характеризується повільнішим прогресуванням шкірно-м’язового синдрому, розвитком полісистемного процесу (до одного року), як правило, на тлі субфебрилітету, помірної або мінімальної запальної активності. Рідко патологічний процес поширюється на внутрішні органи. При такій формі перебігу можлива спонтанна ремісія. У дітей найчастіше спостерігається саме цей варіант.
Первиннохронічний перебіг ЮДМ характеризується повільним прогресуванням симптомів (протягом декількох років), первинним ураженням шкіри, м’язів, мінімальною вісцеральною патологією та мінімальною запальною активністю крові. Клінічно проявляється незначною м’язовою слабкістю, невираженими міалгіями, які можуть виникати після фізичного навантаження, але спостерігається прогресуюча гіпотрофія м’язів, з’являються кальцинати та контрактури суглобів.
Хронічний перебіг ЮДМ можна діагностувати у хворих, у яких початок захворювання характеризувався як гострий або підгострий, а в подальшому на фоні мінімальної запальної активності та зникнення вісцеральних проявів виявляється прогресування шкірно-м’язового синдрому.
Характеристика ступенів активності ЮДМ наведена в таблиці 2.
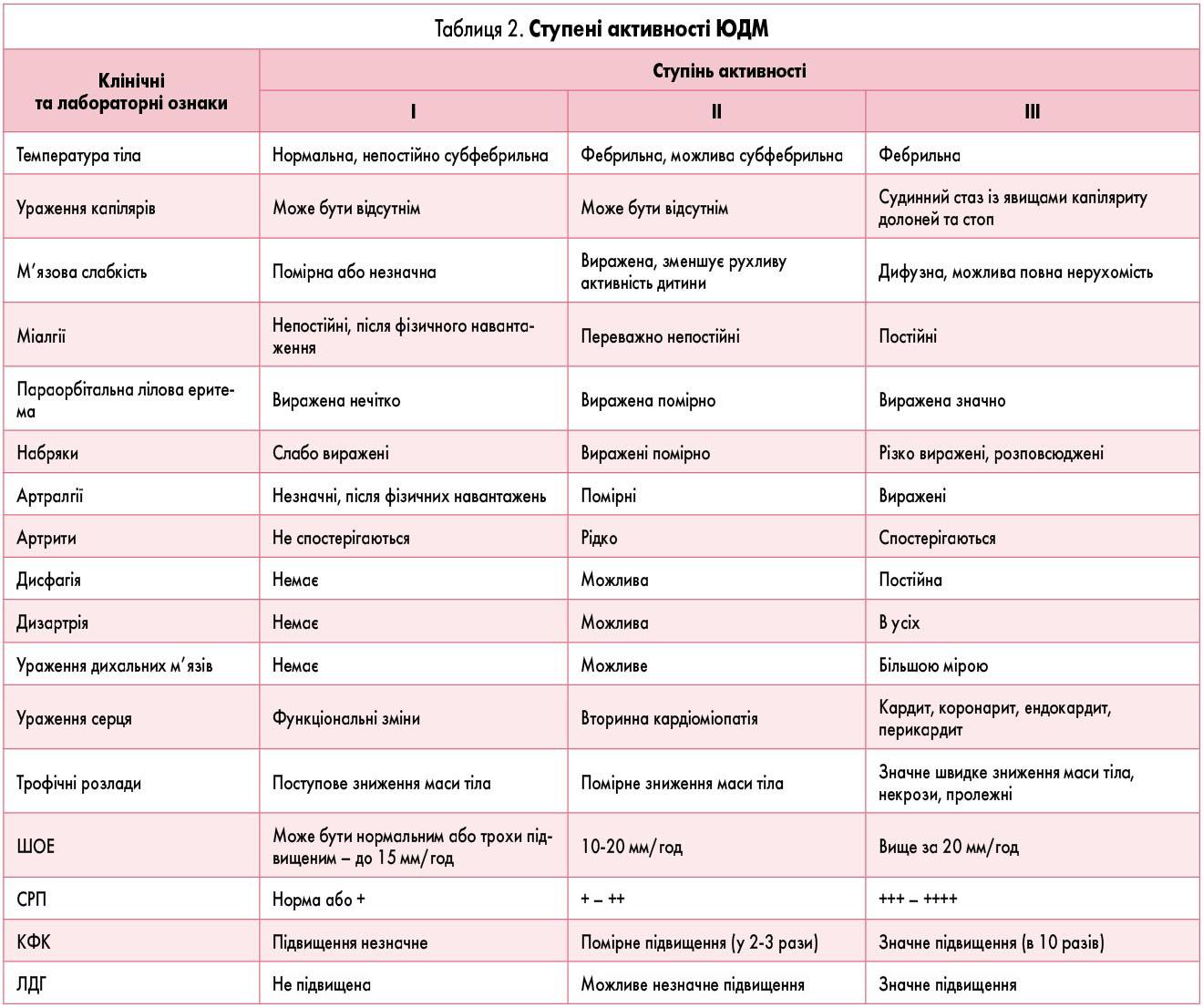
При високому ступені активності та гострому перебігу ЮДМ на початку захворювання у дитини може виникнути міопатичний криз: важке ураження поперечно-смугастої мускулатури із лихоманкою, важка інтоксикація, некротичний панміозит, в тому числі дихальних, глоткових, діафрагмальних м’язів, повна нерухомість, міогенний параліч дихання, що може призвести до смерті пацієнта при відсутності своєчасних реанімаційних заходів.
Неактивна фаза патологічного процесу при ЮДМ характеризується повною клініко-лабораторною ремісією після відміни глюкокортикоїдної терапії. Взагалі при гострому та підгострому перебігу захворювання ця фаза наступає через 2,5-3 роки лікування, при хронічному – через 4-6 років, при первиннохронічному – через 6-9 років.
Для діагностики ЮДМ можна використовувати діагностичні критерії, надані Tanimoto та спіавт. (1995).
Шкіряні критерії:
- пурпурно-червона набрякова еритема верхньої повіки;
- ознака Готтрона: пурпурно-червона атрофічна еритема із лущенням на розгинальних поверхнях суглобів пальців;
- еритема шкіри розгинальної поверхні суглобів кінцівок.
Критерії поліміозиту:
- слабкість у проксимальних групах м’язів верхніх, нижніх кінцівок та тулуба;
- підвищення рівня сироваткової креатинфосфакінази (КФК);
- спонтанний м’язовий біль;
- зміни на електроміограмі – поліфазні потенціали малої тривалості, спонтанні фібриляції;
- позитивний тест на антигістиділ-тРНК-синтетазні антитіла;
- недеструктивні артрити та артралгії;
- ознаки системного запалення (лихоманка, підвищення ШОЕ, позитивний СРП);
- інформативні дані матеріалу біопсії (запальна інфільтрація скелетних м’язів із дегенерацією та некрозом м’язових фібрил, ознаки активного фагоцитозу та регенерації).
При наявності одного чи більше шкірного критерію та не менш ніж 4-х критеріїв поліміозиту можна ставити діагноз ЮДМ.
Клінічна характеристика ЮДМ
Початок захворювання гострий, з явищами м’язової слабкості, яка прогресує, болю в уражених м’язах, зі значним зменшенням ваги тіла, лихоманкою. При первиннохронічному перебігу м’язова слабкість збільшується протягом декількох місяців або навіть років. Іноді першими симптомами хвороби бувають поліартралгії, синдром Рейно.
Ведуча ознака захворювання – ураження м’язів:
- симетрична слабкість проксимальних груп м’язів верхніх та нижніх кінцівок, шиї (неможливість підвести голову від подушки, неможливість встати без сторонньої допомоги, важкість при підйомі по східцях, при піднятті рук тощо). При найбільш важкому перебігу спостерігається ураження всіх м’язів скелетної мускулатури, включаючи м’язи, що беруть участь у диханні та ковтанні;
- ураження м’язів глотки, стравоходу, горла (дисфонія, утруднення при ковтанні, кашель);
- болючість м’язів, особливо вранці та при пальпації;
- при тривалому перебігу захворювання – м’язова атрофія, зменшення маси тіла.
У більш ніж 50% хворих за період перебігу хвороби від 1 до 5 років спостерігається кальциноз м’язів та підшкірної клітковини, можливе виникнення свищів. Вогнища кальцинозу утворюються внаслідок звапніння ділянок шкіри, підшкірної клітковини, м’язів, тканини окремих органів. Звапніння – складний процес, розвитку якого сприяє зміна білкових колоїдів і рівня pH крові, порушення регуляції рівня кальцію в крові, місцеві ферментативні (наприклад, активація фосфатаз) і неферментативні (наприклад, переважання лужного середовища тканини) чинники. Звапнінню передують підвищення метаболічної активності клітин, збільшення синтезу ДНК і РНК, білка, хондроїтинсульфатів, а також активація ряду ферментних систем.
При ЮДМ провідну роль у розвитку кальцинозу відіграють місцеві порушення метаболізму в самій шкірі або підшкірній жировій клітковині, м’язах. Зміни сполучної тканини, судин шкіри та підшкірної клітковини обумовлюють фізико-хімічну спорідненість тканини до солей кальцію. Припускають, що в результаті ацидотичних зрушень тканин знижується парціальний тиск двоокису вуглецю і зменшується розчинність кальцію, що сприяє його відкладенню.
Кальциноз може бути обмеженим, поширеним або універсальним, з відкладенням солей не тільки в шкірі, але й у м’язах, сухожильних піхвах. У випадках обмеженого кальцинозу шкіри тверді вузлики виникають переважно на верхніх кінцівках, в першу чергу в області суглобів; рідше уражаються нижні кінцівки, вушні раковини. При універсальній формі вузли різної величини з'являються на інших ділянках тіла (наприклад, на спині, сідницях). Шкіра, що покриває вузли, спаюється з ними, іноді стає тонкою і проривається. При цьому з вузла виділяється молочно-біла крихкоподібна або кашоподібна маса. Це так звані кальцієві гуми, які можуть сформувати свищі, що відрізняються млявістю течії і вкрай повільним загоєнням. У важких випадках великі суглоби стають нерухомими, відбувається атрофія відповідних груп м’язів, процес супроводжується лихоманкою, кахексією та може призвести до смерті пацієнта.
Синдром васкуліту характеризується сітчастим і деревовидним ліведо (останнє характерне для дітей молодшого віку), долонним капіляритом, гіперемією нігтьового ложа і локалізується у плечовому та тазовому поясі, у проксимальних відділах кінцівок. Можлива поява на шкірі і слизових оболонках трофічних порушень у вигляді виразок і некрозів.
Ураження шкіри. Шкірні прояви ЮДМ у більшості випадків з’являються на декілька місяців раніше ураження м’язів. Вони бувають наступними:
- пурпурно-червона набрякова еритема на верхніх повіках, вилицях, крилах носу, верхній губі. Найбільш часто має вигляд параорбітальної еритеми у вигляді окулярів, або полумаски, може розповсюджуватися на волосисту частину голови. Еритема уражає відкриті ділянки тіла;
- папульозні, бульозні елементи висипу, пурпура, телеангіоектазії, гіперкератоз, пойкілодермія;
- ознака Готтрона: пурпурно-червона атрофічна еритема з лущенням на розгинальних поверхнях суглобів пальців;
- еритема шкіри розгинальної поверхні суглобів кінцівок;
- почервоніння, лущення шкіри долонь;
- навколонігтьова еритема у вигляді червоної кайми з гіпертрофією кутикул;
- фотодерматоз;
- свербіж шкіри;
- алопеція.
Ураження слизових оболонок:
- виразки, гіперемія слизової рота;
- гіперемія, крововиливи у кон’юнктиви очей;
- набряк верхніх дихальних шляхів.
Ураження суглобів:
- симетричне ураження дрібних суглобів, найчастіше суглобів кистей рук, лучезап’ястних, з можливим розвитком м’язових контрактур, але без ерозивних змін у кістках; на фоні тривалого перебігу може сформуватися стійка деформація суглобів.
Ураження серця:
- симптоми міокардиту (помірне розширення меж серця, приглушеність тонів, артеріальна гіпотензія, зміни зубця Т, зміщення інтервалу ST донизу від ізолінії на ЕКГ;
- можливе виникнення аритмій та порушення провідності;
- можуть уражатися всі оболонки серця (ендокард, міокард, перикард), коронарні судини, навіть із розвитком інфаркту міокарда.
Ураження нервової системи:
- переважно периферійної та вегетативної.
Ураження легенів:
- ураження діафрагмальних м’язів із розвитком задишки експіраторного типу;
- гострий дифузний альвеоліт із швидким розвитком легеневої недостатності;
- гіповентиляція (ураження м’язів міжребер’я, діафрагми), аспірація при порушенні ковтання;
- розвиток аспіраційної пневмонії – найбільш важке ускладнення, яке призводить до смерті пацієнта;
- гіпостатична пневмонія внаслідок ураження м’язів, які беруть участь у диханні;
- при високому ступені активності аутоімунного процесу може виникнути плеврит із ексудативним компонентом.
Ураження шлунково-кишкового тракту:
- дисфагія, біль у животі, закрепи;
- езофагіт, гатрит, гастроентероколіт;
- виразка шлунка;
- псевдоабдомінальний синдром, який спостерігається на фоні ураження м’язів передньої черевної стінки і може проявлятися болем, який посилюється під час пальпації;
- в окремих особливо тяжких випадках спостерігається оклюзія судин, які живлять кишечник, що може бути причиною абдомінальних проблем; кальцифікати навкруги м’язових волокон, у місцях, які підлягають механічним впливам (плечові, ліктьові, колінні суглоби, спина). Кальцифікати розташовуються підшкірно або внутрішньошкірно біля уражених м’язів. Якщо вони знаходяться глибоко, виявити їх можливо тільки під час рентгенологічного обстеження. Кальцифікати найчастіше спостерігаються у хворих з яскравими проявами синдрому васкуліту з рецидивуючим перебігом захворювання.
Особливості лабораторних досліджень:
- ознаки запального процесу (підвищення рівню ШОЕ, позитивний СРП);
- підвищений рівень ферментів «м’язового розпаду» КФК, лактатдегідрогенази (ЛДГ), аспартатамінотрансферази (АСТ);
- підвищення рівня антинуклеарного фактору сироватки крові (неспецифічний показник);
- тест на гістиділ-тРНК-синтетазні антитіла (анти-Jo‑1) є позитивним переважно при ураженні легень, а також при розповсюдженому поліміозиті;
- підвищений рівень анти-SRP (антитіла до саркоплазмотичного ретикулюма) виявляється при поліміозиті (відсутності ураження шкіри) та ураженнях серця;
- підвищений рівень антитіл до білково-ядерного комплексу із невизначеною функцією (анти-Мі‑2) характерний для дерматоміозиту; зустрічається у 12% хворих, метод визначення є високоспецифічним (100%), але малочутливим (4-18%);
- РФ може бути присутнім у 10% хворих.
Особливості інструментальних досліджень:
- електроміографія (поліфазні потенціали малої тривалості, спонтанні фібриляції, зниження амплітуди біонапруги уражених м’язів);
- ЕКГ (ознаки порушення метаболічних процесів у міокарді, порушення ритму та провідності);
- ЕХО-КГ (розширення порожнин серця, потовщення та ізоехогенність стінок, зниження скоротливої функції міокарду);
- УЗІ м’язів (порушення архітектоніки м’язових волокон);
- рентгенологічне обстеження (виявлення кальцифікатів);
- КТ грудної клітки виявляє посилення локального або тотального судинного малюнка. При розвитку інтерстиціального легеневого запалення спостерігаються легеневі інфільтрати, легеневий фіброз (базальний або дифузний), субплевральні порожнини (булли), а у разі їх розриву – рентгенологічна картина пневмотораксу;
- МРТ є надійним інструментом для оцінки запалення м’язів під час діагностики та може допомогти диференціювати активну і неактивну стадію захворювання під час спостереження;
- м’язову біопсію доцільно проводити у гострому запальному періоді без явищ атрофії (запальна мононуклеарна інфільтрація скелетних м’язів із дегенерацією та некрозом м’язових фібрил, ознаки активного фагоцитозу та регенерації, ознаки васкулопатії з ураженням ендотеліальних клітин). Дистрофія м’язових структур характеризується розволокненням, втратою поперечної посмугованості, потоншенням. Запальна реакція проявляється периваскулярним набряком, вогнищами інфільтрації лімфоцитів та плазматичних клітин.
Диференційний діагноз
ЮДМ треба диференціювати від великої кількості захворювань, особливо в початковому періоді, коли симптоми бувають нечіткі та дуже споріднені із ювенільним поліміозитом (при ньому відсутні шкірні зміни, а діагноз виставляється за результатами біопсії), міозитом при інших системних захворюваннях сполучної тканини, інфекційним міозитом (виявляються ознаки інфекції, наявність гельмінтозу), нейром’язовими захворюваннями та міопатіями (огляд та обстеження у невролога), онкологічними захворюваннями (МРТ), ендокринопатіями (огляд ендокринолога), міопатіями, які викликані лікарськими препаратами та токсинами.
При нейром’язових захворюваннях та міопатіях:
- часто приєднується випадіння рефлексів і атрофія м’язів;
- виявляється тільки руховий розлад;
- порушення чутливості відсутні;
- ураження майже завжди симетричне (виняток становить міастенія).
Міопатії, викликані лікарськими препаратами та токсинами, можуть виникати під час використання ГК, хлорохіну (делагілу), гідроксихлорохіну (плаквенілу), тривалому вживанні гормонів щитовидної залози у високих дозах. Для стероїдної міопатії характерні нормальний рівень КФК, збільшення м’язової сили після зниження дози ГК.
Лікування ЮДМ
- Обов’язковий стаціонарний етап для встановлення клінічного діагнозу, призначення стартової терапії, контролю показників запальної активності.
- Дієта, що містить багато білка, поліненасичених жирних кислот, кальцію і вітаміну D для профілактики остеопорозу та обмежує вживання вуглеводів, солі. При псевдобульбарних розладах (поперхування) годувати пацієнта повинен хтось з медичного персоналу, щоб уникнути аспірації. У важких випадках показане встановлення назогастрального зонду.
- Етичні проблеми: потрібні повна довіра та розуміння пацієнта, його рідних для контролю та оцінки стану дитини, ефективності довготривалого лікування, можливості зміни лікарських препаратів. Ліки призначаються тільки перорально.
- Протипоказання: вакцинація, введення гамаглобуліну, інсоляція, зміна клімату, переохолодження (в тому числі купання у водоймах), фізичні та психічні травми, контакти з домашніми тваринами, лікування імуномодуляторами в разі розвитку гострої респіраторної інфекції.
- ГК – преднізолон, метилпреднізолон (1-2 мг/кг на добу після преднізолону) в залежності від об’єму уражених м’язів, органів, активності запального процесу на 1-3 міс. Добова доза розподіляється на 3 прийоми, за відсутності позитивної динаміки протягом 2-х тижнів доза збільшується. За необхідності швидкого впливу преднізолон можна вводити внутрішньовенно. Доза вважається адекватною, якщо протягом 4-х тижнів помітно зменшується м’язова слабкість, спостерігається зниження КФК, показників запальної активності крові. Клінічний ефект преднізолону при ЮДМ розвивається повільно, тому максимальну дозу треба призначати на 2 місяці. Після досягнення терапевтичного ефекту доза преднізолону зменшується поступово (на 1/4 від загальної дози, тобто на 2,5 мг на тиждень, наступна 1/4 від загальної дози – 1,25 мг на тиждень), під контролем показників рівнів КФК, ШОЕ, СРБ, самопочуття хворого. Подальше зменшення дози преднізолону проводять індивідуально, дуже повільно до підтримуючої дози (5-10 мг на добу). Тривалість прийому преднізолону повинна становити 3-5 років. Відміняти препарат можна лише під час клініко-лабораторної ремісії протягом року або більше. Якщо активність запального процесу висока, необхідно посилити лікування за допомогою пульс-терапії метилпреднізолоном, цитостатичними препаратами, внутрішньовенним імуноглобуліном. Допускається повна відміна преднізолону у період повної клінічної ремісії або навпаки підвищення дози при загостренні захворювання. Для профілактики ураження слизової оболонки шлунка або кишечнику препарат треба приймати після їжі, запиваючи відваром насіння льону або вівса. Побічні дії преднізолону також включають затримку росту, ризик розвитку інфекційних захворювань, стероїдного остеопорозу, підвищення артеріального тиску.
При міопатичному кризі, високій активності (III ступінь) ЮДМ почати терапію необхідно із пульс-терапії метилпреднізолоном (10-15 мг/кг) від 3 до 5 введень кожен день або через день. - Цитостатичні препарати: метотрексат призначається у дозі 10-15 мг/м2 на тиждень, тільки перорально. Ефект розвивається через 1-2 міс лікування. Тривалість прийому становить 1-3 роки до досягнення стійкої клініко-лабораторної ремісії та за умови відсутності ускладнень. Для зменшення токсичного впливу метотрексату на організм призначається фолієва кислота у дозі 1 мг на добу, окрім днів прийому метотрексату. Основною побічною дією метотрексату є нудота, виразки у порожнині рота, випадіння волосся, підйом рівня аланінамінотрансферази. Циклоспорин А призначається пацієнтам у дозі 5 мг/кг на добу перший місяць, далі – підтримуюча доза 2 мг/кг на добу довготривало, азатіоприн (імуран) – 2-3 мг/кг на добу, підтримуюча доза становить 20-50 мг на добу 2-3 роки.
При міопатичному криз, високій активності ЮДМ (III ступінь) із ураженням легенів призначається циклофосфамід у дозі 10 мг/кг 1 раз на місяць внутрішньовенно на період 6-12 місяців, потім 1 раз на 3 місяця на період 6-12 місяців, далі – 1 раз на 6 місяців до досягнення стійкої клініко-лабораторно-інструментальної ремісії. - Мікофенолат мофетилу при тяжкому, резистентному до стандартної терапії, ураженні шкіри призначається в дозі 600 мг/м2 поверхні тіла 2 рази на добу, мікофенолова кислота – в дозі 450 мг/м2 2 рази на добу.
- За неефективності лікування ГК і лікування імуносупресорами протягом 3-х місяців, при високій активності хвороби (II-III ступінь, міопатичний криз), поширеному шкірному синдромі, вираженому васкуліті, розвитку життєзагрозливих станів (дисфагія, міокардит, інтерстиціальна пневмонія, дихальна недостатність) застосовують імунобіологічні препарати, зокрема химерні моноклональні антитіла до CD20+ В-лімфоцитів – ритуксимаб. Препарат вводять внутрішньовенно в дозі 375 мг/м2 на тиждень протягом 4 послідовних тижнів. Повторний курс проводять через 22-24 тижні після першого введення препарату, якщо зберігається висока активність хвороби. При призначенні ритуксимабу показане призначення ко-тримоксазолу (триметоприму) перорально в профілактичній дозі 5 мг/кг на добу під час лікування ритуксимабом, а також протягом року після його відміни для профілактики пневмоцистної інфекції.
- Встановлений кальциноз може реагувати на лікування бісфосфонатами (памідронат, алендронат), інфліксімабом, абатацептом, внутрішньовенним імуноглобуліном. Можлива хірургічна резекція при рецидивуючому бактеріальному ускладненні.
- Цінним додатком до імуносупресивної терапії при лікуванні ЮДМ є внутрішньовенний імуноглобулін, який показаний хворим із резистентністю до вищеозначеної терапії, важкою дисфагією, при приєднанні бактеріальної інфекції (2 г/кг маси тіла 1 раз на місяць або 1 г/кг за 2 дні протягом 3-6 міс). Внутрішньовенний імуноглобулін пригнічує запальний процес та має імунорегулюючу дію.
- НПЗП при больовому м’язовому та суглобовому синдромі призначаються епізодично для зняття болю: диклофенак натрію – в добовій дозі 2-3 мг/кг маси тіла на 3 прийоми, ібупрофен – 10-15 мг/кг на добу за 3-4 прийоми, німесулід (дітям старше 12 років) – у дозі 1,5-5 мг/кг 2 рази на день після їжі, мелоксикам – у дозі 7,5 мг на добу, призначається дітям від 12 років та старше на тривалий час.
- Препарати, які покращують метаболізм в уражених м’язах: мілдронат (250 мг 1-3 рази на добу протягом місяця), кардонат (дітям 5-15 років по 1 капсулі 2 рази на добу), рибоксин (по 0,2 мг 2-3 рази на день протягом місяця), ретаболіл (0,5-1 мл 5% розчину 1 раз на місяць № 2-3).
- Для покращення кровообігу у м’язах та внутрішніх органах призначають судинні препарати (пентоксифілін – спочатку крапельно по 20 мг на рік життя за два прийоми, при покращенні стану – перорально в тій же дозі на 12 міс або дипіридамол 5 мг/кг на добу до 12 міс.
- Лікувальна фізкультура та масаж підбираються індивідуально, відповідно до об’єму ураження та ступеня активності. Лікар-реабілітолог навчає дітей та їх батьків комплексу вправ лікувальної фізкультури, які направлені на укріплення м’язів, підвищення їх активності при фізичному навантаженні, підвищення об’єму руху суглобів.
Ускладнення:
- остеопороз кісток, особливо хребта, внаслідок значного зниження рухової активності дитини (а також у результаті терапії ГК) з розвитком компресійних переломів і корінцевого синдрому;
- інфікування та нагноєння кальцинатів;
- некрози шкіри;
- аспіраційна пневмонія та асфіксія внаслідок аспірації їжі або блювотних мас;
- дихальна недостатність внаслідок вираженої м’язової слабкості, ураження легень;
- серцева недостатність.
Прогноз захворювання залежить від форми, важкості перебігу, залучення у патологічний процес життєво важливих органів, наявності ранньої адекватної терапії. При її своєчасному призначенні позитивний прогноз спостерігається у 90% хворих.
Диспансерне спостереження за пацієнтом проводиться в умовах поліклініки, із контролем за лікувальними заходами, призначеними в стаціонарі. Необхідний відповідний режим: індивідуальне навчання для виключення стресових ситуацій, виключення інсоляції, звільнення від щеплень, лікувальна фізкультура, легкий масаж, санаторно-курортне лікування. За необхідності проводиться своєчасна планова госпіталізація для контролю стану хворого та корекції лікування. Огляд кардіоревматолога повинен здійснюватися 1 раз у 2-3 місяці з вивченням клініки, рівня КФК, показників запальної активності крові. З диспансерного обліку дитина з ЮДМ не знімається.
Тематичний номер «Педіатрія» №3 (50), 2019 р.