


20 грудня, 2019
Спостереження протягом 12 років за застосуванням ферментозамісної терапії у рідних брата і сестри з ослабленою формою мукополісахаридозу 1 типу: важлива роль початку лікування на ранньому етапі
Мукополісахаридоз 1 типу (МПС‑1) – це захворювання з аутосомно-рецесивним типом успадкування, спричинене дефіцитом α-L-ідуронідази, що характеризується прогресуючим перебігом із залученням у патологічний процес багатьох систем організму: ураженням очей, серця, шлунково-кишкового тракту, легень, скелета і нервової системи. Хоча існує значна клінічна гетерогенність, пацієнтів можна розподілити на 2 категорії, що охоплюють 2 основних клінічних фенотипи: з тяжкою формою захворювання, також відомою як синдром Гурлер, і ослабленою формою хвороби, що раніше називали синдромом Гурлер/Шеє і синдромом Шеє [1].
Синдром Гурлер проявляється в ранньому дитячому віці та характеризується раннім і прогресуючим ураженням багатьох систем організму, включаючи нервову, і за відсутності лікування призводить до летального кінця протягом першого десятиліття життя. У пацієнтів з ослабленою формою МПС‑1 відмічається значна клінічна гетерогенність; симптоми проявляються найчастіше в середньому або у старшому дитячому віці, але встановлення остаточного діагнозу може затриматися до другого десятиліття життя. У хворих з ослабленою формою МПС відбувається повільніше, але безперервне його прогресування, що охоплює багато систем організму, без наявності або з наявністю легких первинних уражень центральної нервової системи. Очікувана тривалість життя пацієнтів з нелікованою ослабленою формою МПС‑1 становить від двох десятків років до середньої тривалості життя.
Ферментозамісна терапія (ФЗТ) ларонідазою, рекомбінантною формою α-L-ідуронідази людини (Альдуразим®*), стала стандартом лікування пацієнтів з ослабленою формою МПС‑1 [2]. У цій статті представляємо дані
Клінічний випадок
Рідні брат і сестра
Брату і сестрі у цей час 12 і 17 років відповідно. Сестрі діагностували МПС‑1 у віці 5 років, у цей час у неї були класичні ознаки ослабленої форми МПС‑1. Завдяки встановленню діагнозу сестрі її брату діагноз було встановлено незабаром після народження. Детальні дані про випадок наведені у попередній публікації [3]. Брат і сестра є носіями складних гетерозиготних мутацій IDUA (W402X і L535F). В обох дітей щорічно проводили повні клінічні та біохімічні дослідження. Кожній дитині розпочали щотижневі інфузії ларонідази в дозі 0,5 мг/кг маси тіла у віці 5 місяців (брат) і 5 років (сестра). Інфузії виконували в лікарні з використанням імплантованих пристроїв венозного доступу; вони добре переносилися пацієнтами, побічних реакцій не зареєстровано; крім того, було пропущено дуже мало інфузій.
Брат
 Пацієнт Ч. (рис. 1); маса тіла 56,0 кг (>97-го процентилю), зріст 168,0 см (>97-го процентилю), об’єм голови (ОГ) 57,2 см (98-й процентиль). З віку 2 років у цієї дитини відмічався зріст 97-го процентилю. Зріст батьків: батька – 170,0 см (20-й процентиль), матері – 165,0 см (75-й процентиль).
Пацієнт Ч. (рис. 1); маса тіла 56,0 кг (>97-го процентилю), зріст 168,0 см (>97-го процентилю), об’єм голови (ОГ) 57,2 см (98-й процентиль). З віку 2 років у цієї дитини відмічався зріст 97-го процентилю. Зріст батьків: батька – 170,0 см (20-й процентиль), матері – 165,0 см (75-й процентиль).
У віці 12 років у цього хлопчика не сформувалися риси обличчя, характерні для МПС‑1. Розвиток його інтелекту нормальний (IQ 116), а якість життя (згідно з оцінкою за EQ‑5D-Y [4]) оцінюється як 100/100; не повідомлялося про порушення дихання або сну; при клінічному обстеженні гепатоспленомегалії не виявлено.
У результаті оцінювання опорно-рухового апарату було зафіксовано легке обмеження згинання-розгинання зап’ястка (60/30; нормальне значення 75/70), надп’ятково-гомілкового суглоба та 4-го і 5-го пальців білатерально. Діапазон рухів у плечовому і кульшовому суглобах нормальний, ознак сколіозу немає.
Показники формальної оцінки слуху є нормальними. Під час офтальмологічних обстежень в 12-місячному віці було виявлено помутніння рогівки легкого ступеня, що залишилося незмінним. З 7 років у пацієнта розвинулися далекозорість та астигматизм легкого ступеня, що були повністю скориговані за допомогою лінз.
За допомогою ехокардіографії у 7 років вперше виявлено ознаки потовщення мітрального і трикуспідального клапанів з недостатністю легкого ступеня. Дилатація передсердя легкого ступеня була визначена до 9 років, але досі залишається практично незмінною – клас I за класифікацією Нью-Йоркської асоціації серця (NYHA).
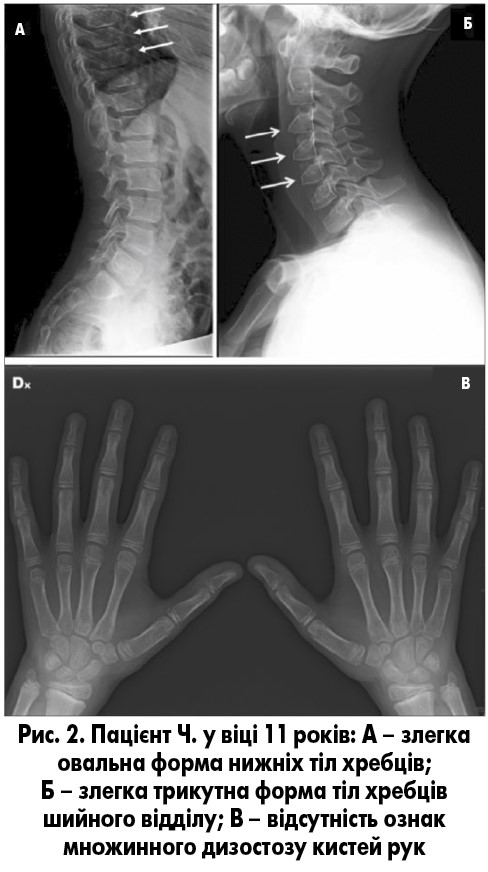 Рентгенографія скелета у віці 10 років продемонструвала наявність мінімального множинного дизостозу нижніх тіл хребців грудного відділу (рис. 2А) і трикутну форму деяких тіл хребців шийного відділу (рис. 2Б); рентгенограми кистей були нормальними (рис. 2В), практично без змін. Дані остеоденситометрії нормальні.
Рентгенографія скелета у віці 10 років продемонструвала наявність мінімального множинного дизостозу нижніх тіл хребців грудного відділу (рис. 2А) і трикутну форму деяких тіл хребців шийного відділу (рис. 2Б); рентгенограми кистей були нормальними (рис. 2В), практично без змін. Дані остеоденситометрії нормальні.
Результати електронейрографії свідчать про наявність білатеральних легких ознак ураження серединного нерва. Головний мозок за даними магнітно-резонансної томографії – в нормі. Не зареєстровано рецидивних інфекцій. Результати перевірки слухових викликаних потенціалів стовбура головного мозку (ABR) та аудіометрії перебувають у межах нормальних значень.
Загальні рівні екскреції глікозаміногліканів (ГАГ) із сечею були підвищені до проведення ФЗТ і нормалізувалися через 4 місяці терапії [3]. Вміст ГАГ у зразках, взятих безпосередньо перед інфузією, залишався в межах нормальних вікових значень до 8,5 року, коли підвищився до 89 мкг/cr (нормальне значення 37±18 мкг/cr) до досягнення 10,5 року, а далі нормалізувався. Результати електрофорезу ГАГ у сечі, що до проведення ФЗТ характеризувалися наявністю дерматансульфату (ДС) і гепарансульфату (ГС), нормалізувалися після 4 місяців ФЗТ до віку 6 років, коли співвідношення вмісту ДС і хондроїтин сульфату (ХС) склало 50/50 та залишається таким до теперішнього часу.
Рівень ГАГ у плазмі крові у віці 11,5 року становив 2,8 мкг/мл (нормальне значення 2,7±1,12 мкг/мл) зі співвідношенням ХС/ДС 98/2 (норма 100/0); вони подібні до результатів, отриманих у віці 6, 9 і 10 років. До проведення ФЗТ не був доступним зразок плазми крові. Після 12 років ФЗТ циркулювальні антитіла до ларонідази при використанні методу імуноферментного аналізу не виявлялися.
Сестра
Пацієнтка Ж. (рис. 1); маса тіла 58,0 кг (>50-го процентилю), зріст 154,0 см (10-й процентиль), ОГ 59 см
Результати обстеження опорно-рухового апарату вказують на прогресування ураження суглобів з обмеженням діапазону рухів як у проксимальних, так і в дистальних суглобах. У пацієнтки розвинулися контрактури всіх пальців (деформація у вигляді кігтеподібної кисті) і тяжке обмеження діапазону рухів у всіх суглобах, зокрема, в ліктьових, колінних і надп’ятково-гомілкових суглобах відмічалося фіксоване згинання 15, 20 і 15° відповідно; хребет – прямий. Пацієнтка не може повністю підняти руки над головою.
Під час офтальмологічного обстеження виявлено виражене дифузне помутніння рогівки, наявне з віку встановлення діагнозу; ступінь помутніння залишався незмінним після 12 років терапії. У віці 11 років у пацієнтки розвинулися далекозорість та астигматизм, що було повністю скориговано за допомогою лінз (гострота зору 10/10).
Через раніше наявний персистуючий рецидивний середній отит пацієнтка має легку двобічну кондуктивну приглухуватість, яка персистувала у 16 років, що було підтверджено шляхом ABR та аудіометрії. Не відмічаються порушення дихання або сну.
Під час дослідження серця не було виявлено значного прогресування помірної недостатності мітрального клапана з потовщеними стулками клапана, про що повідомлялося раніше, пролапсу переднього краю і потовщення лівого передсердя легкого ступеня (II клас за NYHA).
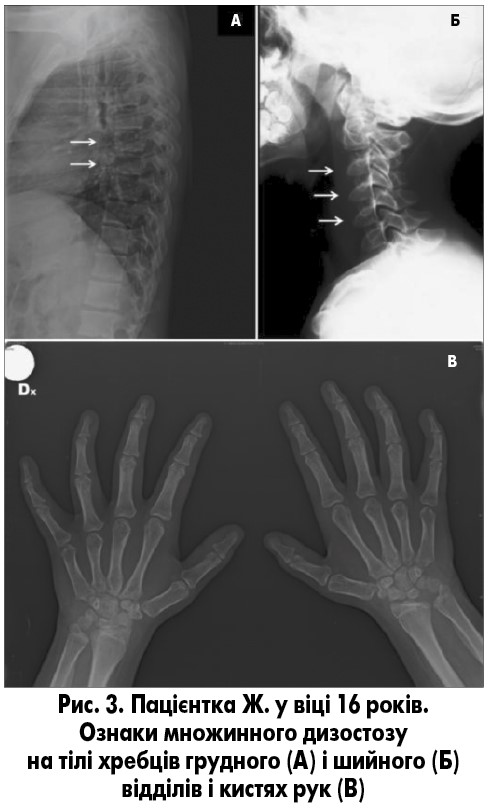 Під час рентгенографії кісток скелета не зафіксовано зменшення вираженості помірно тяжкого множинного дизостозу з ознаками незначного погіршення змін з боку грудних хребців, але відмічалися стабільні результати щодо шийних хребців і кистей рук (рис. 3). Ознаки множинного дизостозу були у дівчинки вже у віці 4,5 року [3]. Результати остеоденситометрії нормальні для її віку. У зв’язку з прогресуванням труднощів ходьби у 16 років пацієнтці було проведено білатеральну елонгацію ахіллового сухожилка.
Під час рентгенографії кісток скелета не зафіксовано зменшення вираженості помірно тяжкого множинного дизостозу з ознаками незначного погіршення змін з боку грудних хребців, але відмічалися стабільні результати щодо шийних хребців і кистей рук (рис. 3). Ознаки множинного дизостозу були у дівчинки вже у віці 4,5 року [3]. Результати остеоденситометрії нормальні для її віку. У зв’язку з прогресуванням труднощів ходьби у 16 років пацієнтці було проведено білатеральну елонгацію ахіллового сухожилка.
Результати спірометрії продемонстрували змішаний вентиляційний патерн (функціональна життєва ємність легень – 66%, об’єм форсованого видиху за 1 с – 70%, пікова швидкість видиху – 71%, максимальна об’ємна
Загальний рівень екскреції ГАГ із сечею був незначно вищим за верхню межу норми для віку пацієнтки протягом останніх 6 років. Електрофорез ГАГ у сечі дав можливість визначити співвідношення ДС і ХС 50/50, що вже спостерігалося у віці 11 років [5]. ГС, наявний до проведення ФЗТ, зник через 5 місяців.
Рівень ГАГ у плазмі крові у віці 5 років до проведення ФЗТ становив 18,2 мкг/мл (нормальне значення 5,27±3,01 мкг/мл) зі співвідношенням ХС/ДС 25/75 (нормальне значення 100/0); через 6, 9 і 10 років ФЗТ рівні ГАГ в плазмі залишалися в межах вікової норми, лише зі слідовими кількостями ДС (співвідношення ХС/ДС становило 98/2). Після 12 років ФЗТ рівні циркулювальних антитіл до ларонідази не були виявлені.
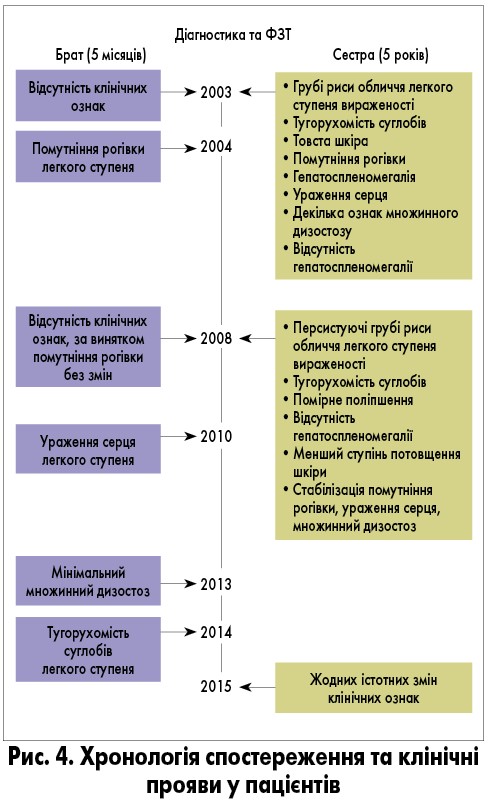 Хронологію спостереження та прояви захворювання наведено на рис. 4.
Хронологію спостереження та прояви захворювання наведено на рис. 4.
Висновки
Сотні пацієнтів з МПС‑1 отримували ФЗТ з часу схвалення ларонідази [6-8], однак у більшості з них лікування починали вже за наявності значного тягаря захворювання. Опубліковані дані довгострокового спостереження вказують на те, що хоча ФЗТ здатна істотно змінити природний перебіг захворювання, її спроможність інвертувати основні клінічні прояви обмежена [9-12]. Останнім часом ці результати були також підтверджені N.A. Al-Sannaa та співавт. [13] у 4 пацієнтів, які почали отримувати лікування протягом першого року життя. Справді, ФЗТ може інвертувати деякі клінічні ознаки, такі як товщина шкіри та гепатоспленомегалія, але щодо зворотного розвитку відхилень діапазону рухів у суглобах можливості лікування обмежені. Крім того, ФЗТ є менш ефективною щодо зміни інших проявів (після того, як вони встановлені), таких як множинний дизостоз, патологія серцевого клапана та помутніння рогівки [7; 9], які є результатом складного патогенетичного каскаду, що призводить до незворотного ураження сполучної тканини [11].
Початок ФЗТ при наявності значної артропатії може зумовити незначне поліпшення діапазону рухів у суглобах протягом 1-2 років лікування з подальшою стабілізацією захворювання або повільнішою швидкістю його прогресування. Ці спостереження дали можливість припустити, що дуже ранній початок ФЗТ, тобто до виникнення значних уражень скелета, може забезпечити поліпшення їх наслідків у довгостроковій перспективі. Подібні докази наводяться відносно потенційного впливу раннього початку ФЗТ на інші ключові клінічні ознаки, такі як прояви з боку серця, офтальмологічні та респіраторні прояви.
Порівняльні дані спостереження після 12 років ФЗТ у цих унікальних рідних брата і сестри дозволяють отримати переконливі докази щодо істотних відмінностей у впливі ФЗТ на пацієнтів з МПС‑1, коли терапію розпочинають до виникнення значних проявів захворювання. Слід зазначити, що ураження скелета, суглобів і серцевого клапана продовжували прогресувати протягом 11 років терапії у сестри, у якої лікування було розпочато у 5 років, тоді як у її молодшого брата, у котрого терапію було розпочато у віці 5 місяців, відмічали мінімальні ураження суглобів і серця без ознак прогресування ураження рогівки. Незважаючи на відсутність симптомів ураження скелета у брата, у якого терапію було розпочато в молодшому віці, у нього зафіксовано ознаки ураження грудних хребців, а також клапанів серця у віці до 7 років. Результати біохімічних досліджень, що проводилися у брата і сестри, свідчать про те, що на більш пізньому етапі в дитинстві спостерігалися помірне збільшення загальних рівнів ГАГ у сечі і більш виражене – ДС. Крім того, загальний рівень ГАГ у плазмі крові був нормальний, лише зі слідовими кількостями ДС. Ні в сечі, ні в плазмі крові не виявлено ГС до початку ФЗТ. Нещодавно De Ru та співавт. [14] і Langereis та співавт. [15] повідомляли, що наявність ДС і ГС може бути пов’язана з дозою препаратів для ФЗТ або статусом антитіл у пацієнтів. На підставі цих даних, а також результатів клінічних і біохімічних досліджень наших пацієнтів ми припустили, що кількість ферменту може бути однією з причин недостатньої деградації ГАГ. Таким чином, поява клінічних ознак навіть після початку терапії на ранньому етапі означатиме, що слід розглянути можливість або збільшення дози ФЗТ, або застосування додаткових ад’ювантних терапевтичних засобів. Дослідження на моделях МПС у тварин свідчать про те, що ранні зміни позаклітинного матриксу та запальні механізми, ймовірно, відіграють певну роль у прогресуванні ураження скелета і сполучної тканини [16; 17]. Таким чином, може знадобитися додавання ад’ювантної терапії, спрямованої на ці метаболічні шляхи, або терапії, орієнтованої на зниження субстрату, для забезпечення кращого результату для пацієнтів.
Цей клінічний випадок показує, що ранній початок терапії істотно змінює природний перебіг ослабленої форми МПС‑1, а також демонструє важливість і потенційний вплив, які неонатальний скринінг і ранній початок терапії можуть мати при наявності цього прогресуючого захворювання.
Список літератури знаходиться в редакції.
Стаття друкується у скороченні.
Gabrielli et al. BMC Medical Genetics. 2016; 17: 19.
* Лікарський засіб Альдуразим®, концентрат для приготування розчину для інфузій, 100 ОД/мл, зареєстрований в Україні. Р.п. № UA/8093/01/01. Наказ МОЗ України № 1449 від 03.08.2018. Зміни внесено наказом МОЗ № 2205 від 31.10.2019.
Тематичний номер «Педіатрія» №4 (51), 2019 р.