


21 грудня, 2019
Сучасні погляди на механізми дії метформіну
На відміну від багатьох препаратів метформін отримано з природного матеріалу, який здавна використовувався у фітотерапії, а не розроблено з орієнтацією на певний шлях чи механізм хвороби. Попри клінічне застосування впродовж 60 років молекулярні механізми дії метформіну залишаються дискусійними.
Метформін і печінка
Гіпоглікемічний ефект метформіну реалізується через вплив на печінку. Так, у мишей із дефіцитом транспортера органічних катіонів 1 типу (ОКТ1) порушується його надходження в печінку та не знижується глікемія за умов годування їжею з високим умістом жиру. У людей метформін знижує утворення глюкози печінкою з мінімальним впливом на периферійне інсулінзалежне поглинання глюкози. Численні дослідження гепатоцитів мишей і трансгенних мишей вказують на здатність метформіну знижувати печінковий глюконеогенез та/або інсулінорезистентність.
Метформін і мітохондріальний контроль печінкового глюконеогенезу
У процесі глюконеогенезу на кожну молекулу синтезованої глюкози витрачається 6 еквівалентів АТФ. Баланс між потребою та витратами АТФ забезпечується мітохондріями. Оскільки метформін несе позитивний заряд, а зовнішні трансмембранні потенціали позитивні, він легко проникає в цитоплазму та мітохондрії, де його концентрації у 1000 разів вищі, ніж у позаклітинному середовищі. У мітохондріях метформін пригнічує комплекс I дихального ланцюга й утворення АТФ. Як наслідок, змінюється відношення АДФ/АТФ у клітинах, де відбувається глюконеогенез.
Іншим наслідком гальмування дихального ланцюга є зміни співвідношення НАД+/НАДГ. Ендогенне утворення глюкози (ЕУГ) печінкою гальмується лише через 1 год внутрішньовенного введення метформіну щурам, супроводжується збільшенням відношення лактат/піруват, що вказує на проблему з повторним окисненням цитоплазматичної НАДГ. Однією із систем, які переносять відновні потенціали із цитоплазми в мітохондрії для повторного окиснення, є гліцерофосфатний човник. Встановлено, що метформін пригнічує його ключовий компонент – мітохондріальну гліцерофосфатдегідрогеназу (мГФД). Введення олігонуклеотидів, які зменшують чутливість мГФД, знижує ЕУГ та скасовує гіпоглікемічний вплив метформіну.
Отже, механізмом гіпоглікемічної дії метформіну є пригнічення мГФД та комплексу I. Можливі також інші впливи на властивості мембрани, а також на взаємодії й окиснення іонів міді, зв’язаних з амінокислотами.
Молекулярні механізми активації AMПK
Пригнічення функції мітохондрій пояснює також здатність метформіну активувати клітинний енергетичний сенсор – АМФ-активовану протеїнкіназу (AMПK). Збільшення відношень АМФ/АТФ та АДФ/АТФ активує АМПК, яка відновлює енергетичний гомеостаз, перемикаючись на катаболічні шляхи, що генерують АТФ і вимикають АТФ-витратні клітинні процеси, тобто синтез клітинних запасів поживних речовин перемикається на їх розпад.
Під впливом бігуанідів збільшується клітинне відношення AМФ/АТФ та АДФ/АТФ, оскільки AMПK не активується ні метформіном, ні фенформіном у клітинах, які експресують мутантну AMПK, нечутливу до змін AMФ або AДФ. Однак активація AMПK можлива також унаслідок глюкозного голодування та дії низьких концентрацій метформіну. У цьому випадку вона реалізується за іншим механізмом – шляхом утворення комплексу з лізосомальним білком LAMTOR1 (активатор 1 mTOR). Отже, активація АМПК під впливом метформіну може відбуватися не лише через мітохондрії, а й через лізосоми.
AMПK-залежний та AMПK-незалежний впливи на печінковий глюконеогенез
Очевидно, що деякі гострі ефекти метформіну на продукцію печінкової глюкози не залежать від AMПK. Зокрема, це пригнічення фруктозо-1,6-бісфосфатази AMФ.
Основним довготривалим клінічно значимим ефектом метформіну є посилення чутливості печінки до інсуліну. Вважається, що цей процес опосередкований AMПK, яка швидко пригнічує синтез та активує окиснення жиру в печінці шляхом прямого фосфорилювання двох ізоформ ацетил-КoA-карбоксилази (АКК1/АКК2) в еквівалентних залишках серину. У мишей основної групи, у яких обидва залишки серину було замінено на залишки аланіну, що не фосфорилюються (AКК1-S79A та AКК2-S212A), рівні діацил- та тріацилгліцерину в печінці та м’язах були підвищені внаслідок посилення синтезу жиру та зменшення його окиснення. Крім того, в тварин спостерігалися стеатоз, гіперглікемія, гіперінсулінемія, порушення толерантності до глюкози (ПТГ) і резистентність до інсуліну навіть за умов нормальної дієти. Коли мишей із групи контролю на 6 тиж переводили на дієту з високим умістом жиру, в них виникали такі ж гіперглікемія та ПТГ, як і в мишей основної групи. Проте після 6 тиж лікування метформіном ці показники значно покращувалися, натомість у мишей основної групи – не змінювалися.
Отже, метформін підвищує чутливість до інсуліну шляхом фосфорилювання AКК1 та AКК2. Оскільки фосфорилювання АКК скасовується нокаутом AMПK, очевидно, що довгострокові сенсибілізуючі інсулінові ефекти метформіну цілком залежні від AMПK.
Метформін і кишечник
Препарат збільшує анаеробний метаболізм глюкози в ентероцитах, зменшує кінцеве захоплення глюкози та посилює надходження лактату в печінку. Генетичні дослідження в людини встановили, що втрата функції гена, який кодує ОКТ1 (SLC22A1), знижує надходження метформіну в печінку, але не впливає на його здатність зменшувати рівень глікованого гемоглобіну у хворих на цукровий діабет 2 типу. Крім того, в осіб із діабетом форма з уповільненим вивільненням, яка тривало затримується в кишечнику і має мінімальне системне всмоктування, так само ефективно знижує рівень глікемії, як і форма негайного вивільнення.
Є низка можливих механізмів кишкового впливу метформіну на метаболізм глюкози. Зокрема, препарат збільшує утилізацію глюкози в кишечнику; цей ефект підтверджено позитронно-емісійною томографією: значне поглинання фтордеоксиглюкози (ФДГ), особливо в товстій кишці, з помітним посиленням після 30 днів лікування, що зберігалося впродовж 48 год після відміни препарату. Збільшення поглинання ФДГ супроводжувалося посиленням фосфорилювання AMПK. Обидва ефекти спостерігалися лише в ентероцитах товстої кишки, де глюкоза в просвіті майже відсутня; це дозволяє припустити, що метформін збільшує її захоплення та системний метаболізм.
Іншим механізмом впливу є посилення секреції глюкагоноподібного пептиду-1 (ГПП-1), притаманне для форм як із негайним, так і зі сповільненим вивільненням.
У дванадцятипалій кишці щурів метформін пригнічує продукцію глюкози в печінці через ядро солітарного тракту, вагусні еференті волокна й активацію AMПK та ГПП-1 рецепторів (зв’язок «кишка – мозок – печінка») (рис.).
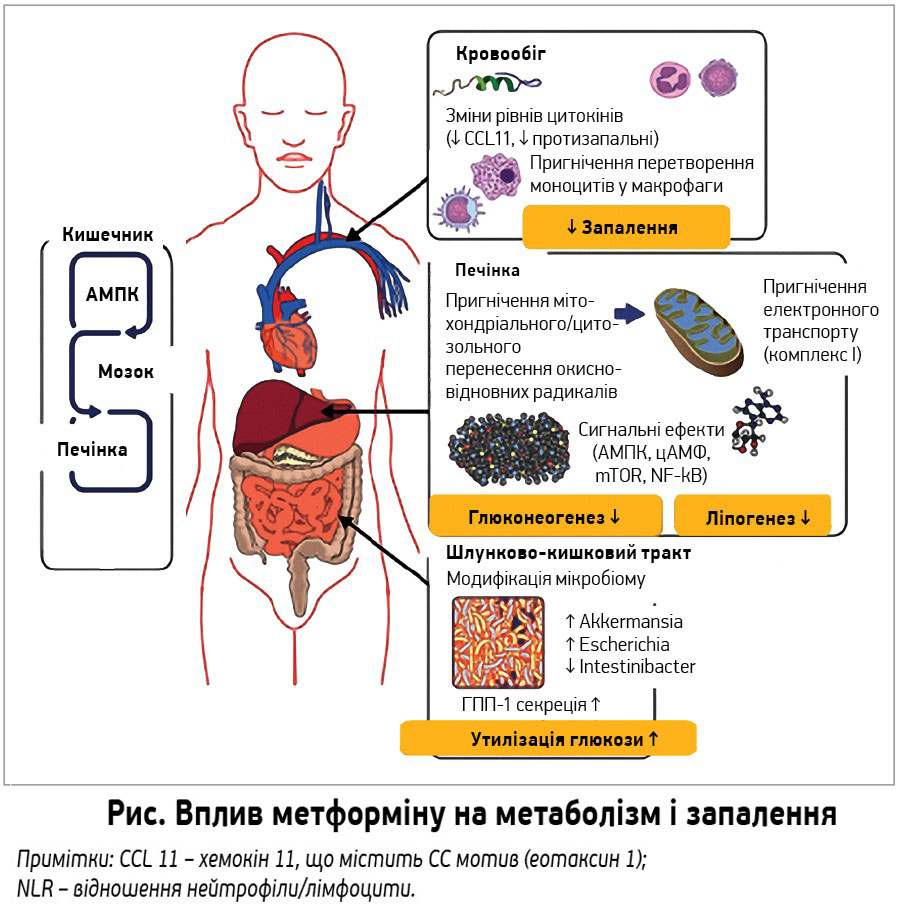
Іншим потенційним механізм кишкової дії метформіну є вплив на мікробіом кишечнику.
Запалення, старіння та вплив на мікробом
Метформін подовжує тривалість життя в нематод Caenorhabditis elegans. Удослідженнях на тваринах під впливом метформіну збільшувалася популяція бактерій виду Akkermansia в кишечнику, що супроводжувалося зменшенням запалення в жировій тканині та зниженням постпрандіальної гіперглікемії. Унещодавніх дослідженнях мікробіому кишечнику в людей із різних країн встановлено, що найчастіше метформін збільшує види Escherichia і зменшує види Intestinibacter. Відтак, зміни мікробіому при цукровому діабеті 2 типу пов’язані з метформіном, а не з діабетом, хоча їх причинно-наслідкове значення в терапевтичній користі метформіну потребує подальшого вивчення.
Метформін прямо впливає на запалення, зокрема на передачу сигналів NF-κB, зменшує перетворення моноцитів у макрофаги та пригнічує синтез прозапальних цитокінів; у хворих на цукровий діабет 2 типу зменшує відношення нейтрофілів до лімфоцитів – запальний маркер, що підвищує ризик смерті та серцевих подій. Одним із таких цитокінів є еотаксин-1, який сприяє віковій клітинній і тканинній дисфункції. Можливо, саме цим пояснюється здатність метформіну продовжувати тривалість життя ссавців. Іншими потенційними механізмами такого впливу є АМПК-залежні та незалежні механізми регуляції сигнального шляху mTOR – мішені рапаміцину ссавців.
Отже, метформін прямо або опосередковано впливає на печінку, знижуючи утворення глюкози. Впливаючи на кишечник, він посилює утилізацію глюкози, збільшує ГПП-1 та змінює мікробіом. На молекулярному рівні метформін пригнічує мітохондріальний дихальний ланцюг у печінці, активуючи AMПK, посилює чутливість до інсуліну через вплив на жировий обмін та знижує цАМФ, зменшуючи тим самим експресію глюконеогенних ферментів. Можливий також AMПK-незалежний вплив на печінку – пригнічення фруктозо-1,6-бісфосфатази AMФ. Ці механізми потребують подальшого вивчення та мають бути перевірені в людини за умов орального застосування препарату.
Стаття друкується в скороченні.
Rena G., Hardie G., Pearson E.R. The mechanisms of action of metformin. Diabetologia 2017; 60: 1577-1585. DOI 10.1007/s00125-017-4342-z
Переклала з англ. Ольга Королюк
Медична газета «Здоров’я України 21 сторіччя» № 22 (467), листопад 2019 р.