


1 серпня, 2015
Принципы современного пересмотра классификации ихтиозов

Характерным признаком современных клинических направлений является пересмотр нозологических определений, перестановка некоторых заболеваний из одной группы в другую. Такие, казалось бы, неожиданные шаги имеют в своей основе научные исследования последних лет, в основном генетические и иммунологические. Эти довольно кардинальные перестановки уже очевидны во взглядах на заболевания соединительной ткани, «ретикулезы»-лимфомы, токсикодермии, фотодерматозы и др. Претерпела изменения и классификация ихтиозов.
В руководствах по дерматологии начала ХХ в. существовала классификация ихтиозов, преимущественно основанная на клинических вариантах этой патологии. Так, в монографиях таких корифеев дерматологии, как Дарье (1915), Никольский (1918), Григорьев (1940), первоначальная классификация ихтиозов (ichthyosis) преподносилась следующим образом:
- xerosis;
- ichthyosis simplex;
- ichthyosis nitida;
- ichthyosis serpentina;
- ichthyosis hystrix;
- ichthyosis sauriasis.
Практически в основу классификации был положен принцип оценки плотности гиперкератотических чешуек.
В 60-е годы ХХ ст. появилась новая классификация, откликнувшаяся на стремительное развитие медицинской генетики. В соответствии с этой классификацией были вычленены ненаследственные формы ихтиоза, а наследственные – в основном учитывали способ наследования. Таким образом, данная классификация была построена следующим образом:
- Наследственные формы:
- аутосомно-доминантный обычный, эпидермолитический ихтиоз (буллезная ихтиозиформная эритродермия Брока);
- Х-сцепленный рецессивный ихтиоз;
- аутосомно-рецессивный ихтиоз: сухая ихтиозиформная эритродермия.
- Наследственные синдромы, включающие ихтиоз.
- Ихтиозиформные приобретенные состояния (приобретенный ихтиоз):
- симптоматический ихтиоз (гиповитаминоз А, болезни крови, злокачественные заболевания и др.);
- сенильный ихтиоз.
В это же время в дерматологию стали настойчиво проникать генетические исследования, что заставило конкретизировать передающиеся по наследству дерматозы, выделив их в группу генодерматозов. Систематизация генодерматозов позволила выделить следующие нозологические группы:
- наследственные заболевания кератинизации (ихтиозы, ладонно-подошвенные кератодермии, болезнь Дарье, порокератоз и др.);
- наследственный буллезный эпидермолиз и синдромы хрупкости кожи;
- наследственные расстройства соединительной ткани (cutis laxa, синдром Элерса–Данлоса);
- эктодермальные дисплазии; наследственные болезни волос и ногтей (атрихия, монилетрикс, трихотиодистрофия, гипертрихоз, анонихия, пахионихия, трихоринофалангеальный синдром, синдром желтых ногтей);
- наследственные расстройства пигментации (альбинизм, недержание пигмента);
- наследственные метаболические болезни кожи (болезнь Фабри, энтеропатический акродерматит, порфирия).
Таким образом, ихтиозы вошли в раздел расстройств кератинизации, а в 80-е годы совершенно объективно их выделили как нозологическую группу ихтиозиформных генодерматозов. Такая нозологическая группа позволила ввести в нее разные клинические формы, в основе которых лежат учтенные мотивации путей наследования. Ихтиозиформные генодерматозы разделили на:
- вульгарный ихтиоз;
- вульгарный ихтиоз, сцепленный с полом;
- ихтиозиформный гиперкератоз;
- пластинчатый ихтиоз, ихтиозиформная эритродермия;
- эпидермолитический ихтиоз (врожденная буллезная ихтиозиформная эритродермия);
- гистриксоидный ихтиоз.
Исторические вехи формирования представления об ихтиозах, тяжести поражения кожи, прогнозе, наследовании этой патологии заключались в следующем:
- 1806 г. – Aliber упоминает об аутосомно-доминантном вульгарном ихтиозе.
- ХIХ в. – описание ихтиоза Арлекина; разделение буллезного и небуллезного типов ихтиоза Брока. Брок предложил название «врожденная ихтиозиформная эритродермия».
- 1966 г. – Frost, Van Scott четко представили различие между аутосомно-доминантным «эпидермолитическим гиперкератозом» и аутосомно-рецессивным «ламеллярным ихтиозом».
- 1965 г. – Wells, Kerr выделили Х-сцепленный с полом ихтиоз.
- 1970 г. – Wells, Kerr выделили отдельную нозологию – «дефицит стероидной сульфатазы».
В последние годы именно эта группа дерматозов потребовала пересмотра классификации. Проект классификации ихтиозов стартовал на конференции по ихтиозам в 2007 г., когда была сформирована рабочая группа из 37 экспертов: клиницистов, патологов кожи, генетиков, представлявших 12 стран. Любопытно и современно выглядит привлечение к активной работе общественных организаций: «Интернет-общение для пациентов с ихтиозами и расстройствами кератинизации» (Германия); «Фонд ихтиозов» (США); «Организация больных ихтиозом» (Германия).
В результате пересмотра номенклатуры и классификации наследственных ихтиозов были окончательно сформулированы следующие определения.
Наследственные ихтиозы представляют собой большую клинически и этиологически гетерогенную группу менделевских заболеваний ороговения, полностью вовлекающих кожные покровы.
Ихтиозы и эритрокератодермии – клинически и этиологически гетерогенные группы расстройства ороговения. Ихтиозы (греч. – рыба) характеризуются генерализованным шелушением кожи, при эритрокератодермии же очевидны эритема и гиперкератоз, преимущественно фокальные и без отчетливого шелушения. «Синдромные» и «несиндромные» формы ихтиозов предполагают похожие подгруппы:
- аутосомно-рецессивный врожденный ихтиоз: ихтиоз Арлекина, ламеллярный ихтиоз, врожденная ихтиозиформная эритродермия;
- «кератинопатический ихтиоз»: эпидермолитический ихтиоз, поверхностный эпидермолитический ихтиоз Курта—Маклина.
Установление правильного клинического диагноза ихтиоза является предпосылкой к обоснованному прогнозу, терапевтическому решению и предложению генетического консультирования. В клинической диагностике помогает определение следующих параметров: качество и распространенность шелушения, наличие или отсутствие эритродермии, пузырей, отклонения со стороны придатков. Большинство наследственных ихтиозов и эритрокератодермий присутствуют с рождения и манифестируют в неонатальный период.
Помогают дифференцировать заболевания данной группы клинические признаки, наследственность, структурные, биохимические и молекулярные аномалии. Вульгарный ихтиоз обычно не проявляется клинически при рождении. Терапия при вульгарном ихтиозе является симптоматической и первично помогает дифференцировать его от других ихтиозов. Многие, но не все, ихтиозы хорошо «отвечают» на прием оральных ретиноидов. Лечение состоит из низких доз и титруется в зависимости от реакции на препарат.
Вульгарный ихтиоз – это расстройство ороговения с преимущественной распространенностью 1 на 250 индивидуумов, заболевание наследственное с аутосомно-полудоминантным типом наследования. Умеренный ихтиоз наблюдается вследствие гетерозиготной филаггриновой мутации; тяжелый ихтиоз – мутации в обеих аллелях филаггрина. Дефект в гене филаггрина при вульгарном ихтиозе реализуется в комплексе хромосомы 1q21. Заболевание характеризуется неполной пенетрантностью у гетерозигот и вариабельной экспрессией внутри семьи.
При патогистологическом исследовании почти у половины больных установлен недостаток нормального зернистого слоя, при электронной микроскопии обнаруживается дефицит профилаггрин-содержащих кератогиалиновых гранул в зернистом слое, а биохимически – уменьшение или отсутствие профилаггриновой экспрессии в эпидермисе и в культуре кератиноцитов (при нормальном содержании других протеинов). Увеличение сцепления рогового слоя и формирование чешуек приводит к снижению удерживающих воду аминокислот (по причине катаболизма филаггрина).
Признаки вульгарного ихтиоза обнаруживаются в возрасте 2 лет. Ранее по тяжести поражения кожи и внешнему виду кожи больного выделяли: ксеродерму, змеевидный ихтиоз (ichthyosis serpentinа), ящерицеподобный (ichthyosis sauriasis), напоминающий кожу дикобраза (ichthyosis hystrix). Вульгарный ихтиоз характеризуется образованием чешуек диаметром до 1 см желтовато-серого цвета, плотно прикрепленных к коже. Локализация – разгибательные поверхности конечностей и туловище (рис. 1). Кожа на сгибательных поверхностях конечностей, в паховых и подмышечных областях не изменена.
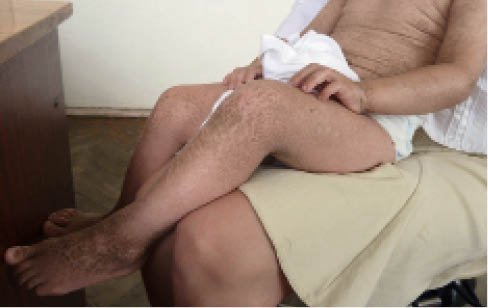 Рис. 1. Клинические проявления вульгарного ихтиоза
Рис. 1. Клинические проявления вульгарного ихтиоза(здесь и далее – фото предоставлены автором)
В соответствии с современной пересмотренной классификацией выделяют клиническую форму дефицит стероидной сульфатазы (синоним: Х-сцепленный рецессивный ихтиоз). Это Х-сцепленное рецессивное заболевание с заболеваемостью 1:2 000–1:5 000. Отсутствие активности стероидной сульфатазы обусловлено делецией STS-гена на хромосоме Xp22.31 у 90% больных. Дефицит стероидной сульфатазы обусловлен низким уровнем или отсутствием эстрогена в моче и амниотической жидкости (неадекватное рассоединение дегидроэпиандростерона).
Этот ихтиоз наблюдается только у мужчин, начинается в первые 6 мес жизни, прогрессирует до пубертатного возраста, затем клиника стабилизируется. Данная форма характеризуется образованием крупных полигональных чешуек, разделенных достаточно широкими трещинами (рис. 2). Обращает на себя внимание очевидное поражение ушей, волосистой части головы и шеи – «болезнь грязной шеи». Отмечено, что достаточно характерно для заболевания улучшение летом, но, вместе с тем, нет улучшения с возрастом. У заболевания есть определенные синдромные стигмы, например, в 100% случаев – офтальмологические расстройства в виде помутнения роговицы, что отмечается и у женщин-носителей генов. У 25% больных – гипогонадизм, может быть крипторхизм, эпилепсия, лимфобластная лимфома. У женщин-носителей наблюдаются осложнения беременности в связи с низким уровнем эстрогенов.
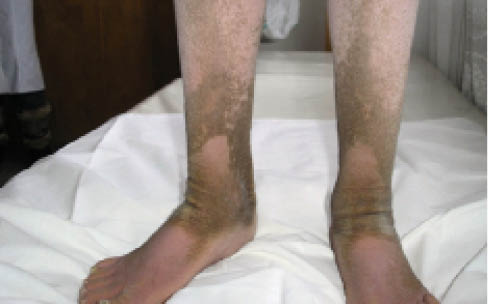 Рис. 2. Клиническая картина дефицита стероидной сульфатазы (Х-сцепленный ихтиоз)
Рис. 2. Клиническая картина дефицита стероидной сульфатазы (Х-сцепленный ихтиоз)Выделяют буллезную врожденную ихтиозиформную эритродермию (синонимы: эпидермолитический гиперкератоз, буллезная врожденная ихтиозиформная эритродермия Брока, буллезный ихтиоз). Гистопатологию первым описал Никольский в 1897 г., а в 1902 г. Брок подробно дифференцировал сухую и буллезную формы. Заболеваемость при буллезной ихтиозиформной эритродермии составляет 1:200 000–1:300 000. Это аутосомно-доминантное заболевание с полной пенетрантностью, нет гендерной зависимости, в 50% случаев возникает спорадически и презентует новые мутации. Возникает вследствие гетерозиготной мутации в генах, кодирующих кератин 1 и кератин 10, локализованных на хромосомах 12q13.3 и 17q21.2. Эпидермальный акантоз и гиперкератоз ведут к гиперпролиферации и уменьшению десквамации, соответственно страдает барьерная функция кожи, что ведет к повышению трансэпидермальной потери воды (TEWL). При рождении кожа новорожденного напоминает обожженную с распространенными пузырями и эрозиями. В младенческий период при трении и давлении возникают вялые пузыри. Со временем характерно появление грязных шипоподобных гиперкератотических чешуек, нередко на эритематозном основании, часто – на сгибательных поверхностях (рис. 3), сопровождается ладонно-подошвенной кератодермией.
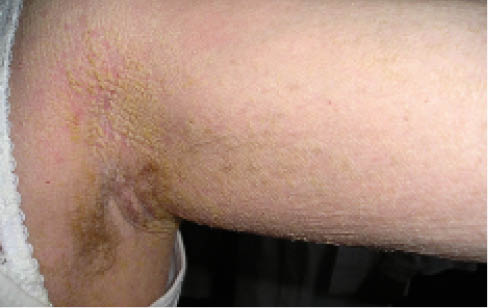 Рис. 3. Клинические проявления буллезной врожденной ихтиозиформной эритродермии
Рис. 3. Клинические проявления буллезной врожденной ихтиозиформной эритродермииОтдельно в этой нозологической группе в настоящей классификации выделяют коллодийный плод. Коллодий – греческое слово, означающее «клей», «липкий». Механизмы формирования коллодийной пленки непонятны и мешают адаптации плода к земной жизни, что достаточно часто приводит к неонатальной летальности. При этой форме ихтиоза установлена мутация в гене трансаминазы-1 на хромосоме 14q12, что ведет к дефициту трансаминазы-1, а это, в свою очередь, нарушает формирование протеина и липидов при ороговении. В результате формируется массивный гиперкератоз, нарушение барьерной функции кожи и TEWL.
Коллодийный плод рождается преждевременно, с высоким риском летальности. При рождении ребенок покрыт блестящей натянутой пленкой (рис. 4). Натянутость приводит к эктропиону, вывороту губ, гипоплазии носа и ушей. Пленка со временем «сбрасывается», и остается нормального вида кожа (пластинчатое отшелушивание новорожденного) либо, более часто, наблюдается одна из форм небуллезной врожденной ихтиозиформной эритродермии или ламеллярного ихтиоза. Оба заболевания гетерогенны и также нередко совпадают. Чрезмерная сухость кожи с TEWL приводит к образованию глубоких трещин, которые легко инфицируются, а это вполне может закончиться сепсисом.
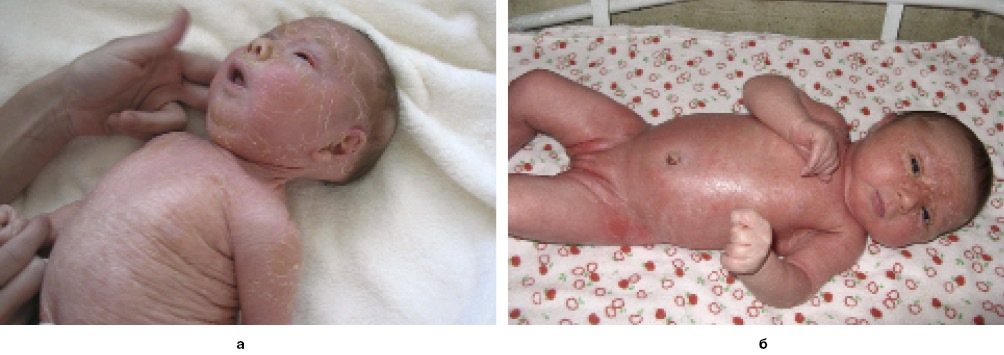 Рис. 4. Клиническая картина коллодийного плода
Рис. 4. Клиническая картина коллодийного плодаВ 1966 г. для всех аутосомно-рецессивных форм небуллезного врожденного ихтиоза введен термин ламеллярный ихтиоз (синоним: пластинчатый ихтиоз). Позже эту группу разделили на 2 фенотипа: ламеллярный ихтиоз и небуллезная врожденная ихтиозиформная эритродермия. Заболеваемость 1:200 000–1:300 000 живых рождений. Любопытно отметить такой изолят с более высокой заболеваемостью (1:91 000), как Норвегию. У большинства пациентов с классической клиникой ламеллярный ихтиоз вызван дефицитом трансаминазы-1.
Аутосомно-рецессивный ламеллярный ихтиоз – одно из наиболее тяжелых заболеваний среди наследственных болезней ороговения, приводит к инвалидности с детства и летальному исходу у новорожденных. Дерматоз существует с рождения в виде яркой эритемы всего кожного покрова с последующим присоединением шелушения крупными полигональными чешуйками (рис. 5, а) или развивается из клинического фенотипа коллодийного плода. Патогномоничными признаками ламеллярного ихтиоза являются симметричность поражения, шелушение кожи в виде плотных полигональных чешуек серовато-коричневого цвета, плотно прикрепленных в центре и отслаивающихся по краям (рис. 5, б), эктропион, деформация ушных раковин и диффузное поражение ладоней и подошв со сглаженностью папиллярных линий на кончиках пальцев рук и ног.
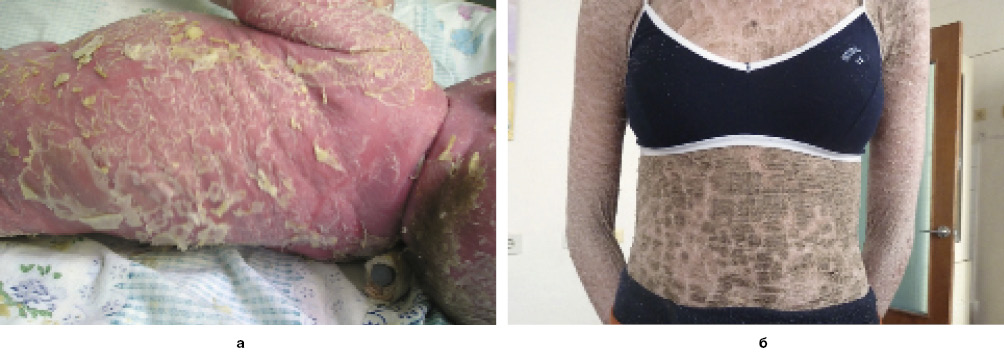 Рис. 5. Клинические проявления ламеллярного ихтиоза
Рис. 5. Клинические проявления ламеллярного ихтиозаК редким формам ихтиоза относят: буллезный ихтиоз Сименса (синоним: эксфолиативный ихтиоз); ихтиоз гистриксоидный Курта–Маклина; ихтиоз гистрикс (от hystrix – дикобраз); ихтиоз Арлекина; синдром Сьогрен–Ларссона (триада: ихтиоз, ди- или тетраплегия, умственное расстройство); трихотиодистрофия с ихтиозом (с рождения эритродермия, шелушение, обломанные волосы); множественный дефицит сульфатазы.
Клинико-генетическая классификация наследственных ихтиозов (несиндромных форм) предполагает конкретное указание на установленный путь наследования при том или ином заболевании. Так, при вульгарном ихтиозе присутствует аутосомно-полудоминантный вариант наследования, при ламиллярном ихтиозе – аутосомно-рецессивный или Х-сцепленный рецессивный, при ихтиозе Арлекина и врожденной ихтиозиформной эритродермии – аутосомно-рецессивный.
В настоящее время дерматолог должен разбираться в разнообразии форм ихтиозов, поскольку именно от него зависит предположительный диагноз, представление о прогностических перспективах, ориентация семьи на последующее генетическое консультирование, объективный взгляд на предоставление инвалидности. На современном этапе положительный прогноз клинической картины возможен при назначении системных ретиноидов и разнообразной наружной десквамативной терапии.
Литература
1. Суколин Г.И. Клиника наследственных дерматозов. Атлас-справочник. М.; 2014: 311 с.
2. Sterry W., Paus R., Burgdorf W. The ichthyosis. In: Dermatology; Georg Thieme Verlag; 2006: 332–40.
3. Richard G., Ringpfeil F. Ichthyoses, Erythrokeratodermas and Related Disorders. In: Dermatology; MOSBY; 2008: 743–76.
4. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference In Soreze 2009. JEADV; 2012.