


30 вересня, 2015
Хвороба Фабрі з ураженням серця: клінічний випадок та огляд проблеми
Хвороба Фабрі – це рідкісне спадкове Х-зчеплене захворювання, що характеризується дефіцитом ферменту лізосомальної α-галактозидази. Як і інші лізосомальні хвороби накопичення, хвороба Фабрі призводить до розвитку поліорганної патології, має різноманітну картину клінічних проявів, що імітують інші порушення, у тому числі серцево-судинні. З цим пов’язані проблеми діагностики хвороби Фабрі – захворювання або не виявляється зовсім, або діагностується на пізній стадії. Останнім часом використовують патогенетичне лікування хвороби Фабрі – ферментозамісну терапію, яка за умови раннього призначення значно покращує стан пацієнтів, уповільнює прогресування уражень, поліпшує прогноз.
Хвороба Фабрі – друга за частотою виявлення лізосомальна хвороба накопичення
Лізосомальні хвороби накопичення – загальна назва спадкових захворювань, пов’язаних із порушенням функції лізосом, внутрішньоклітинних органел, які здійснюють перетравлення екзогенного матеріалу або відпрацьованих органел клітини за допомогою ферментів. Генетично детерміноване порушення синтезу одного або декількох ферментів лізосом призводить до накопичення в них специфічного субстрату цих ферментів (Parkinson, 1961). Клінічна картина окремих хвороб залежить від того, в яких органах та тканинах відбувається накопичення субстрату. Вони часто мають поліорганний характер та потребують диференційної діагностики у пацієнтів із захворюваннями нервової системи, нирок, ознаками м’язової дистрофії або скелетною дисплазією та деформацією, гепатомегалією, спленомегалією, кардіопатіями (Hopkin, 2008). Відомо більше 70 таких хвороб, серед них хвороба Гоше, Фабрі, Помпе, Німана-Піка, Тея-Сакса, мукополісахаридоз та ін. Найбільш поширеною є хвороба Гоше, яка пов’язана з дефіцитом глюкоцереброзидази та має аутосомно-рецесивний тип успадкування (Weinreb, 2006).
Хвороба Фабрі, відома також як хвороба Андерсона-Фабрі, вперше була описана в 1898 р. Два лікаря – німецький дерматолог Йохансен Фабрі (1860-1930) та англійський хірург Вільям Андерсон (1842-1900) – незалежно один від одного опублікували описи клінічних випадків ангіокератом із супутньою протеїнурією у чоловіків (Mehta, 2006). Пізніше були відмічені спадковий характер хвороби та зв’язок із Х-хромосомою, а потім визначена локалізація мутації в Х-хромосомі. Патогенез хвороби Фабрі пов’язаний із дефіцитом ферменту лізосомальної α-галактозидази А, що призводить до накопичення глоботріаозилцераміду (Gb3) в лізосомах клітин у різних тканинах організму (Linhart, 2007). Раніше жінки вважалися лише носіями, в яких хвороба взагалі не маніфестує або має незначні прояви. Але дані епідеміологічних та клінічних досліджень показали, що у гетерозиготних жінок за наявності дефектного гена хвороба розвивається пізніше, ніж у гемізиготних* чоловіків, може мати меншу вираженість (ураження окремих органів та систем) та більш сприятливий перебіг (Mehta, 2004; Wilcox, 2008). Маніфестація хвороби у гетерозиготних жінок зумовлюється випадковою інактивацією Х-хромосоми у різних тканинах, при цьому не виявлено прямої залежності від співвідношення інактивованих Х-хромосом дикого типу до уражених Х-хромосом (Maier, 2006). У гемізиготних жінок активність α-галактозидази А може бути зниженою або навіть нормальною за наявності клінічних проявів хвороби (Chimenti, 2004).
Хвороба Фабрі належить до рідкісних (орфанних) захворювань. За різними даними, її частота варіює від 1 на 40 тис. до 1 на 117 тис. новонароджених (Desnick, 2007), хоча останнім часом завдяки розширенню арсеналу методів діагностики реєструється значно більша кількість випадків хвороби Фабрі, особливо тих її форм, що маніфестують у пізньому віці (Spada, 2006). За даними реєстру Fabry Outcome Survey (FOS), у середньому у жінок хвороба діагностувалася в 32 роки із затримкою в 19 років від появи симптомів, а у чоловіків – у 27 років із затримкою в 15 років (Sunder-Plassmann, 2006). Диференційну діагностику слід проводити з ревматичними захворюваннями, артритами, фіброміалгіями, дерматоміозитами, хворобою Меньєра, нервово-психічними розладами (Mehta, 2004). У кардіологічній практиці це перш за все гіпертрофічна кардіоміопатія (ГКМП).
На сьогодні існує 2 великі реєстри пацієнтів із хворобою Фабрі – Fabry Outcome Survey (Mehta, 2006) та Fabry Registry (Waldek, 2009). Останній є відкритим міжнародним реєстром, до якого можуть долучатися лікарі з усіх країн світу та реєструвати пацієнтів на базі онлайн-платформи (www.registrynxt.com, 2015). За даними Charrow, у 2013 р. у реєстрі нараховувалося 4482 пацієнти – 2144 чоловіка та 2338 жінок, середній вік яких становив 42 та 45 років відповідно.
Клінічні прояви хвороби Фабрі – ураження різних органів та систем
Патогенез та клінічні прояви хвороби Фабрі пов’язані з накопиченням глоботріаозилцераміду та інших гліколіпідів у лізосомах різних органів та тканин, чим зумовлена різноманітність симптомів. Клінічні прояви можуть бути розділені на класичні, серцеві та ниркові (Rosenfeld, 2009; Mehta, 2006).
У разі класичного перебігу активність α-галактозидази становить <1%. Також за часом появи та вираженістю симптомів розрізняють ранній класичний перебіг хвороби з поліорганними порушеннями та пізній, коли активність α-галактозидази знижена до 5-35%, а прояви виникають у дорослому віці, при цьому можливе ураження однієї або кількох систем організму (Mehta, 2006; Golfomitsos, 2012) (табл. 1).
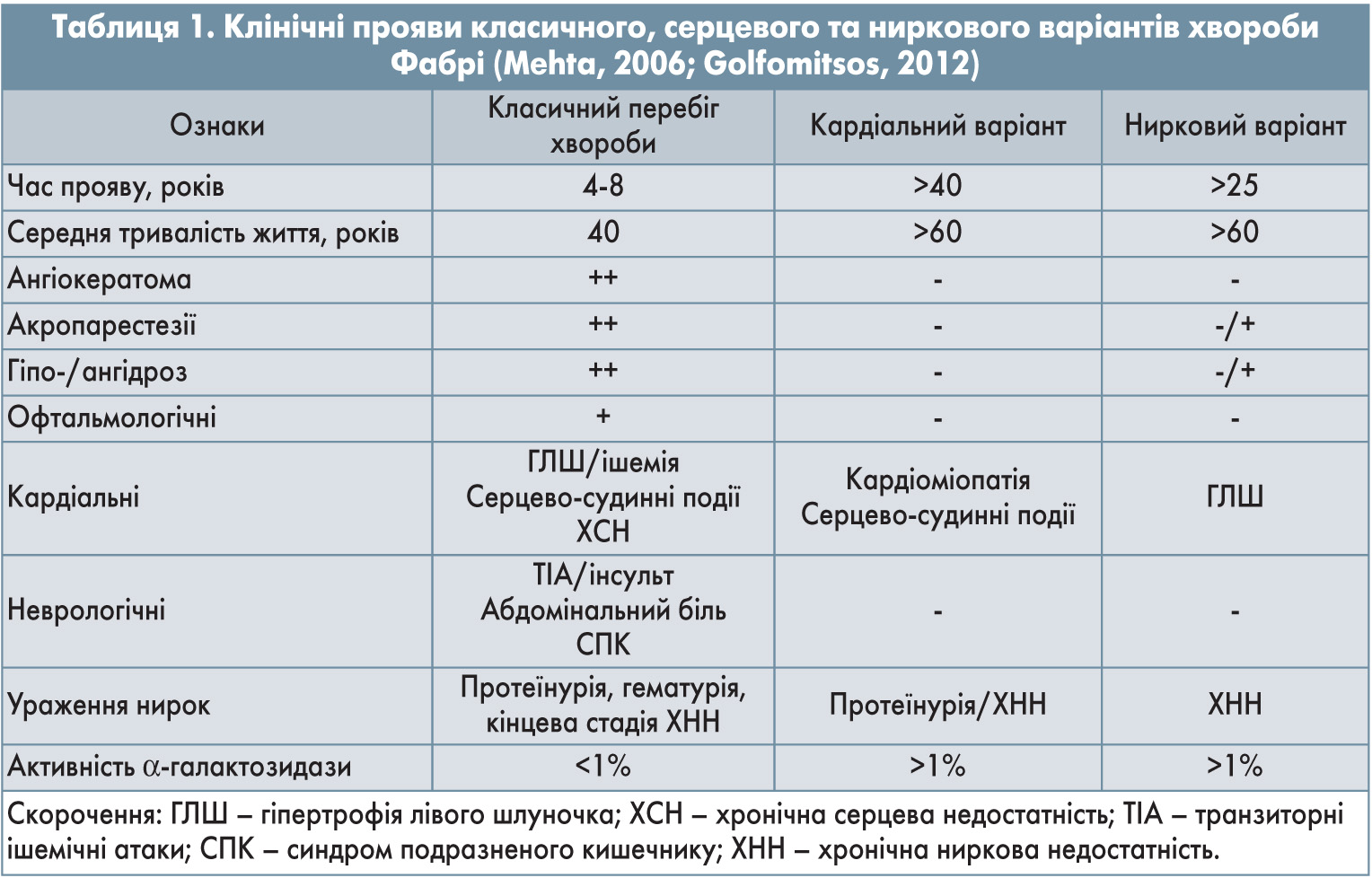
У разі класичного перебігу хвороба маніфестує в дитинстві (5-10 років) у вигляді болю в кінцівках із подальшим розвитком ангідрозу, ангіокератоми, помутніння рогівки та інших симптомів. З віком у хворих можливе виникнення хвороби нирок, ішемічної хвороби серця і цереброваскулярних захворювань, що призводять до передчасної смерті. Нефропатія при хворобі Фабрі зумовлена накопиченням Gb3 у подоцитах ниркових клубочків (Waldek, 2012).
У пацієнтів діагностується гломерулонефрит і, як наслідок, хронічна ниркова недостатність, що на пізніх стадіях потребує гемодіалізу. Дослідження пацієнтів, які знаходяться на гемодіалізі, показали, що до 1,2% осіб мають хворобу Фабрі, яка не була діагностована раніше (Spada, 2006).
З віком симптоми можуть змінюватися, що теж слід ураховувати у разі підозри на хворобу Фабрі. Так, наприклад, акропарестезії можуть бути першим симптомом у дитинстві, який потім безслідно зникає в юнацькому віці (Mehta, 2010). Основні клінічні симптоми та їх розвиток у часі представлені в таблиці 2.

За даними Fabry Outcome Survey (Ramaswami, 2006), найбільш частими ранніми проявами у дітей до 18 років були неврологічні симптоми та розлади з боку шлунково-кишкового тракту, що розвивалися у 80 і 60% пацієнтів відповідно. Неврологічні симптоми включали акропарестезії, генералізований біль, зміни чутливості до температури і дисгідроз. Характерні 2 типи болю – хронічний пекучий біль / акропарестезії та кризи Фабрі, які супроводжуються вираженим пекучим болем у кінцівках, що згодом охоплює інші частини тіла (Germain, 2010). Шлунково-кишкові розлади включали біль у животі, нудоту, діарею або запори. Їх виникнення пов’язують із накопиченням Gb3 у вегетативних гангліях, розташованих у кишечнику. Офтальмологічні порушення, у тому числі cornea verticillata і звивисті судини сітківки, виявлялися у 60% пацієнтів, також у дітей віком до 5 років. Приблизно 40% пацієнтів мали слухові та дерматологічні симптоми, у тому числі шум у вухах, запаморочення та ангіокератоми. У дітей віком >10 років частіше виявлялися мікроальбумінурія та протеїнурія. У дитячому віці можливі кардіальні прояви хвороби Фабрі у вигляді зниження варіабельності серцевого ритму, збільшення маси міокарда порівняно зі здоровими особами (Kampmann, 2008). У хлопчиків порівняно з дівчатами симптоми виникали на 2-5 років раніше (Ramaswami, 2006).
Кардіальні прояви хвороби Фабрі
Ураження серцево-судинної системи при хворобі Фабрі зумовлено як безпосереднім впливом на серце та судини накопичення Gb3 із подальшим розвитком ГЛШ та фіброзу, так і виникненням вторинної артеріальної гіпертензії внаслідок ниркової патології. Накопичення Gb3 спостерігається в кардіоміоцитах, клітинах провідної системи, клітинах ендотелію, гладеньких міоцитах судин серця, фібробластах клапанів (Linhart, 2007). Це призводить до різноманітних змін у вигляді ГЛШ та гіпертрофії правого шлуночка (ПШ), порушень провідності, порушення функції клапанів, мікроваскулярної стенокардії.
Основні клінічні прояви з боку серцево-судинної системи, характерні для хвороби Фабрі, представлені в таблиці 3.
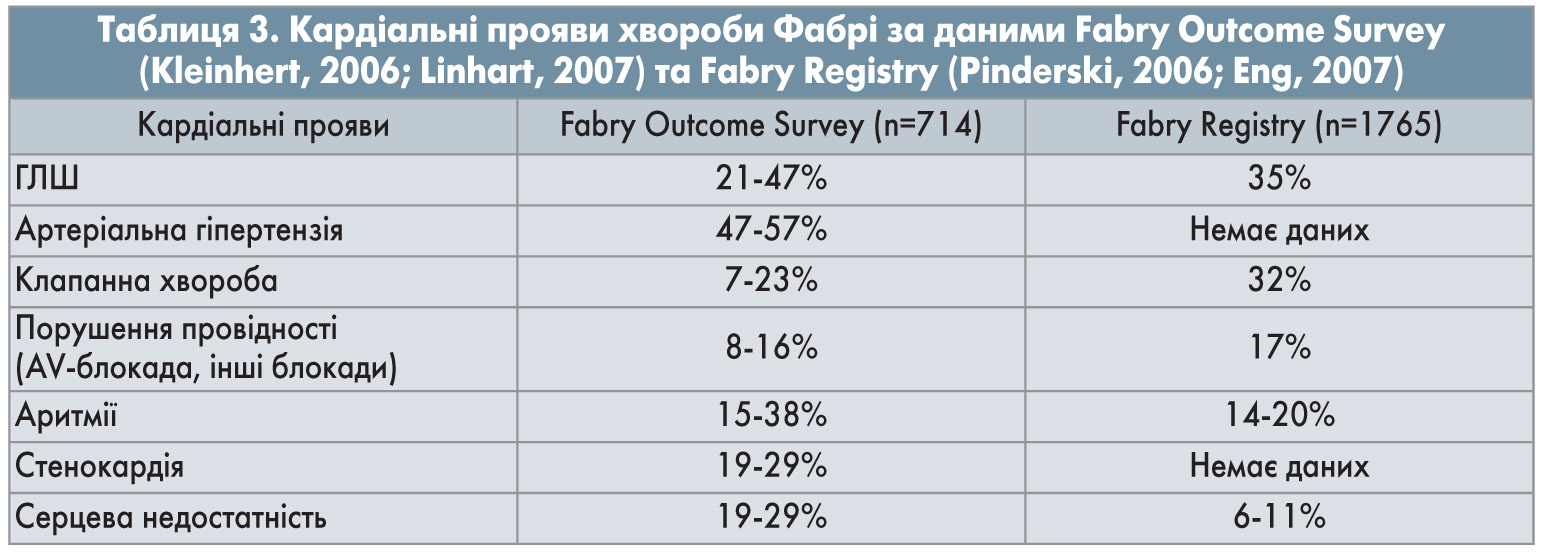
ГЛШ при хворобі Фабрі – основний симптом, який можна легко виявити при проведенні рутинного ехокардіографічного (ЕхоКГ) дослідження; також виявляються характерні зміни на електрокардіограмі (ЕКГ). ГЛШ при хворобі Фабрі зустрічається у 1/2 гемізиготних чоловіків і 1/3 гомозиготних жінок (Kapmann, 2008). При цьому не відбувається дезорганізації міофібрил, а розвиток гіпертрофії пов’язують зі зміною внутрішньоклітинних процесів, що активують патологічні сигнальні шляхи, які, у свою чергу, призводять до гіпертрофії, апоптозу, некрозу та фіброзу (Weidemann, 2013). Також виявлено зв’язок між хронічною хворобою нирок та вираженістю гіпертрофії, артеріальною гіпертензією та серцево-судинними подіями (Talbot, 2015). Потовщення міокарда частіше буває симетричним, але в подальшому спостерігається більш виражений фіброз задньобокової стінки ЛШ, що зумовлює її стоншення відносно міжшлуночкової перегородки (МШП). Іноді розвиток асиметричної ГЛШ при хворобі Фабрі асоціюється з обструкцією виносного тракту ЛШ (Linhart, 2007). Крім фіброзу, на пізніх стадіях захворювання виникає порушення діастолічної та систолічної функцій ЛШ. Можливий розвиток рестриктивної кардіоміопатії. При патології серця також може спостерігатися коронарна недостатність, що пов’язана в основному не з ураженням артерій, а зі збільшенням потреби в кисні, зменшенням щільності капілярів, підвищенням діастолічного тиску в ЛШ та інфільтрацією патологічним субстратом ендотеліальних клітин та м’язового шару артеріол та капілярів (Hulkova, 1999; Linhart, 2006).
Ураження провідної системи серця на ранніх стадіях захворювання характеризується збільшенням швидкості проведення, що визначається скороченням інтервалу PQ (Roudebush, 1973). На більш пізніх стадіях характерні брадикардія, блокада ніжок пучка Гіса та AV-вузла, що може потребувати імплантації штучного водія ритму (Takenaka, 2008). У дослідженні, яке включало 78 пацієнтів із хворобою Фабрі (Shah, 2005), у 4% відмічалася постійна, а у 13% – пароксизмальна форма фібриляції передсердь, у 8% хворих спостерігалася нестійка шлуночкова тахікардія. Як шлуночкова тахікардія, так і брадиаритмії є можливими причинами кардіальної смерті при хворобі Фабрі (Acharya, 2012), тому пацієнтам із кардіальними проявами захворювання рекомендується регулярно проводити добовий моніторинг ЕКГ.
Також можливе ураження клапанів (як правило, мітрального та аортального) у вигляді потовщення та деформації стулок унаслідок відкладання сфінголіпідів та вторинного фіброзу і кальцифікації (Desnick, 1976). Недостатність клапанів частіше незначна або помірна. У реєстрі Fabry Registry серед 1448 пацієнтів у 32% виявлялася мітральна регургітація (Pinderski, 2006).
У реєстрі Fabry Outcome Survey серед 752 пацієнтів у 14,6% спостерігалося ураження клапанів, однак хірургічне втручання було необхідне лише в 3 випадках (Linhart, 2006).
Діагностичний алгоритм у разі підозри на хворобу Фабрі у пацієнта з ГЛШ представлений на рисунку 1.
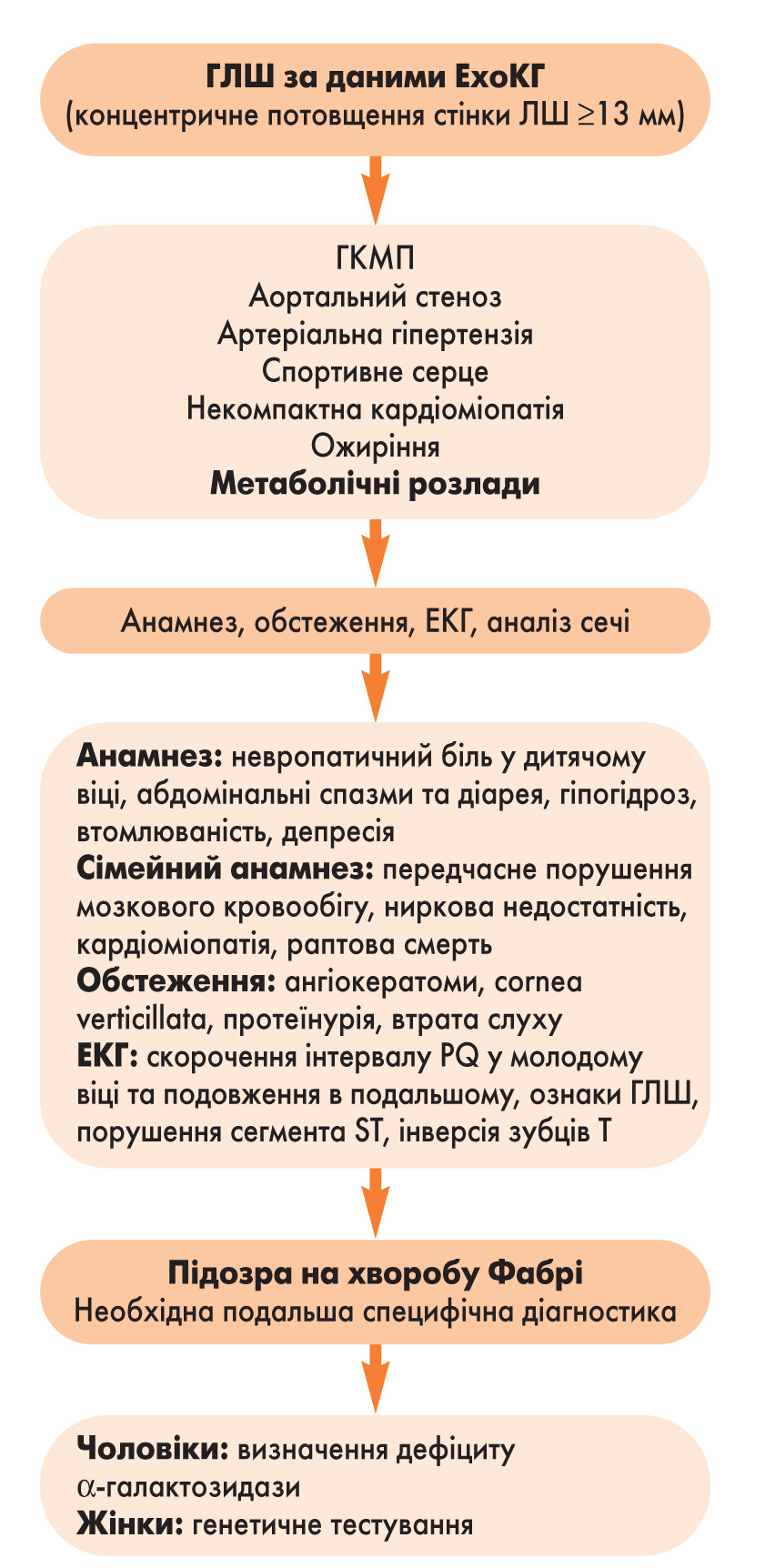 Рис. 1. Діагностичний алгоритм у разі підозри на хворобу Фабрі у пацієнтів із ГЛШ (Yousef, 2013)
Рис. 1. Діагностичний алгоритм у разі підозри на хворобу Фабрі у пацієнтів із ГЛШ (Yousef, 2013)За різними даними, ризик хвороби Фабрі у пацієнтів із ГКМП становить 0,5-12%. Так, у дослідженні FOCUS (Hagege, 2011), яке проводилося у Франції, серед 392 пацієнтів із попереднім діагнозом ГКМП та товщиною стінки ЛШ >1,5 см у 4 (1,5%) чоловіків методом генетичного тестування була виявлена хвороба Фабрі. В іншому дослідженні (Elliot, 2011), що включало 1386 пацієнтів (представників 13 європейських країн) із ознаками ГКМП і товщиною стінки ЛШ >1,5 см, хвороба Фабрі була діагностована у 7 учасників (0,5%; 4 жінки та 3 чоловіка) . У дослідженні за участю 230 пацієнтів із ГЛШ >1,3 см, що проводилося в Японії (Nakao, 1995), хвороба Фабрі була виявлена у 7 (3%) чоловіків. Найвища (12%) частота виявлення хвороби Фабрі зафіксована в італійському дослідженні за участю 34 жінок, в яких ГКМП була виявлена у віці >40 років (Chimenti, 2004). У чоловіків із пізнім виявленням ГЛШ хвороба діагностувалася у 6% випадків (Sadchev, 2002). Пацієнти з артеріальною гіпертензією у більшість досліджень не включалися, що може бути причиною недооцінки поширеності хвороби Фабрі.
Методи кардіальної візуалізації
За рекомендаціями канадської ініціативи щодо хвороби Фабрі (West, 2012), для виявлення ураження серця при діагностованій хворобі Фабрі достатньо наявності двох із нижче наведених критеріїв: стінка ЛШ >12 мм (у чоловіків) та >11 мм (у жінок), гіпертрофія за даними ЕКГ, маса ЛШ на 20% більша норми, рестриктивний тип діастолічної дисфункції ЛШ з E/A >2,0 та час сповільнення потоку раннього діастолічного наповнення (DTE) <140 мс, швидкість збільшення маси ЛШ >5 г/м2 протягом 1 року, порушення стрейну, збiльшення розмiру лівого передсердя, порушення ритму та провiдностi (AV-блокада, скорочення PQ, блокада лівої ніжки пучка Гіса, шлуночковi та суправентрикулярнi тахiаритмiї, синусова брадикардiя), аортальна та мiтральна недостатнiсть (від помірної до вираженої), ознаки фіброзу при проведенні магнітно-резонансної томографії (МРТ) – пізнє підсилення (особливо у жінок за відсутності вираженої гіпертрофії). Кардіоміопатія при хворобі Фабрі, що супроводжується ГЛШ, вважається спадковим варіантом ГКМП, пов’язаним із порушенням метаболізму (Elliot, 2014).
ЕхоКГ-картина при хворобі Фабрі має певні особливості. У хворих частіше розвивається концентрична ГЛШ або концентричне ремоделювання ЛШ (Linhart, 2000). Лише близько 5% випадків гіпертрофії при хворобі Фабрі мають ознаки асиметричної гіпертрофії МШП або верхівкової ГЛШ (Linhart, 2007). Також у більшості випадків гіпертрофований і ПШ. На ранніх стадіях хвороби систолічна функція ЛШ, що визначається двовимірною ЕхоКГ, збережена, але при використанні тканинного допплера можливе зниження систолічних та діастолічних швидкостей, що характеризує порушення скоротливої здатності та розслаблення міокарда (Pieroni, 2006). Також характерним є зниження деформації міокарда (поздовжнього та радіального стрейну) на ранніх стадіях кардіопатії при хворобі Фабрі (Morris, 2014). Рестриктивний тип діастолічної дисфункціїї ЛШ спостерігається на пізніх стадіях хвороби. У дослідженнях у більшості пацієнтів виявлялися незначні порушення діастолічної функції ЛШ, що характеризувалися зменшенням співвідношення Е/А та подовження часу DTE (Pieroni, 2006). Також у деяких пацієнтів відмічається підсилення ехо-сигналу від ендокарда ЛШ, що не характерно для гіпертрофії при ГКМП (Kovacevic-Preradovic, 2008).
При проведенні МРТ серця, окрім виявлення ГЛШ, характерним є наявність ознак фіброзу міокарда ЛШ. Для хвороби Фабрі специфічною локалізацією вважаються базальні задньолатеральні сегменти ЛШ (Moon, 2003; Cobelli, 2009), що визначається на МРТ-зображеннях при пізньому підсиленні гадолінієм. При цьому не відмічається ураження субендокардіального шару, що характерно для інфаркту міокарда, хоча часто реєструються порушення скоротливості у цих ділянках. За наявності ГКМП фіброз міокарда має більш різноманітну локалізацію та може уражати субендокардіальний шар (Cobelli, 2009). Також можливо отримати корисну діагностичну інформацію при МРТ у режимі Т1 до введення контрастної речовини. Цей режим використовується для виявлення жирового компонента у міокарді, що типово для хвороби Фабрі, де субстратом накопичення є сфінголіпіди.
У дослідженнях із використанням методу МРТ було показано, що рівень сигналу Т1 у ділянці МШП у пацієнтів із хворобою Фабрі значно менший, ніж у здорових осіб або пацієнтів із гіпертрофією на тлі артеріальної гіпертензії, при амілоїдозі чи ГКМП (Sado, 2013; Thompson, 2013). При цьому фіброз виявлявся у базальних сегментах задньолатеральної стінки ЛШ. При МРТ-дослідженні у режимі Т2 при хворобі Фабрі відмічається подовження часу релаксації Т2 та підвищення інтенсивності сигналу порівняно з аналогічними параметрами у пацієнтів із ГКМП (Imbriaco, 2007).
Лікування
На сьогодні для патогенетичного лікування хвороби Фабрі використовують 2 препарати – інгібітор α-галактозидази А, або агалзидаза a (Replagal, Shire HGT, США) та інгібітор α-галактозидази Б, або агалзидаза β (Fabrazyme®, Genzyme, США). Досвід їх застосування протягом останніх 10 років свідчить про зменшення темпів прогресування ГЛШ, кращу виживаність пацієнтів, зниження кількості серцево-судинних подій (Germain, 2014). Кращі результати отримано у пацієнтів молодшого віку та при менш вираженому порушенні функції нирок на початку терапії. У дослідженні з прямим порівнянням двох препаратів не встановлено різниці в частоті досягнення таких кінцевих точок, як смерть, серцево-судинні події, кінцева стадія захворювання нирок (S.M. Sirrs et al., 2014).
Лікування кардіальних проявів хвороби Фабрі симптоматичне. З метою терапії артеріальної гіпертензії та нефропротекції рекомендоване застосування інгібіторів ангіотензинперетворюючого ферменту та блокаторів рецепторів ангіотензину (Kleinert, 2006). Також вони показані пацієнтам із ГЛШ без артеріальної гіпертензії. За наявності симптомів стенокардії рекомендовані антиагреганти. β-Блокатори слід призначати з обережністю у зв’язку з можливим розвитком вираженої брадіаритмії та порушення AV-провідності. Існує позитивний досвід використання дигідропіридинових блокаторів кальцієвих каналів (Linhart, 2006). За наявності вираженої брадіаритмії рекомендована імплантація штучного водія ритму, а пацієнтам із загрозливими для життя шлуночковими тахіаритміями – установка кардіовертера-дефібрилятора. Описані випадки проведення алкогольної абляції у пацієнтів із обструкцією виносного тракту ЛШ на тлі хвороби Фабрі (Magage, 2005).
Клінічний випадок
Пацієнтка П., 52 роки, проходила обстеження у ДУ «Національний науковий центр «Інститут кардіології ім. М.Д. Стражеска» НАМН України» у жовтні 2014 р.
Основними скаргами, з якими вона звернулася до лікаря, були прогресуюча слабкість та задишка при помірному фізичному навантаженні. Останніми роками відмічено коливання артеріального тиску з підвищенням до 160/90 мм рт. ст. Порушення з’явилися протягом останнього року та поступово посилювалися. Із анамнезу відомо, що 10 років тому жінці був установлений діагноз ГКМП, необструктивна форма, але вона не спостерігалась у кардіолога та не приймала постійну медикаментозну терапію. На той час її турбувала легка задишка. У хворої також виявлено гломерулонефрит, з приводу якого вона спостерігалась у нефролога протягом останніх 15 років та періодично проходила амбулаторне лікування. Протягом останніх років у жінки відмічалась анемія. У 15-17-річному віці пацієнтку турбував пекучий біль у стопах, який самостійно минув. У 35 років вона народила дитину (хлопчика). У хлопчика у більш ранньому віці (близько 5-6 років) теж з’явився пекучий біль у стопах. Мати помітила схожість симптомів зі своїми скаргами в юнацькому віці, зверталася з метою консультації до різних спеціалістів, у тому числі до психоневролога, але причина болю в дитини не була виявлена. У віці 16 років синові пацієнтки було встановлено діагноз хвороби Фабрі; його підтверджено за допомогою генетичного тестування. Ураховуючи характер успадкування хвороби, скарги та клінічні прояви, жінці також була проведена ферментодіагностика і підтверджена хвороба Фабрі. (Слід зазначити, що це мало місце за 6 міс до обстеження у нашій клініці, тобто на час установлення діагнозу ГКМП 10 років тому хвороба Фабрі ще не була діагностована.) Обстеження пацієнтки виконано в умовах стаціонару закладу. Під час огляду та загальноклінічного обстеження не було виявлено значних відхилень, окрім блідості шкіри. При перкусії ділянки серця визначалося розширення меж відносної серцевої тупості вліво на 1,5 см, при аускультації серця на тлі правильного уповільненого ритму визначався незначний систолічний шум над проекцією аортального клапана, що не проводився на судини.
Діагностика. В аналізах відмічалося помірне зниження рівня гемоглобіну – 109 г/л, збільшення швидкості осідання еритроцитів – 23 мм/год, підвищення рівня С-реактивного білка до 18,07 мг/л та виявлені ознаки ГЛШ, характерні для ГКМП. На ЕКГ – ознаки ГЛШ, характерні для гіпертрофічної кардіопатії, та наявність подовження інтервалу PQ (рис. 2).
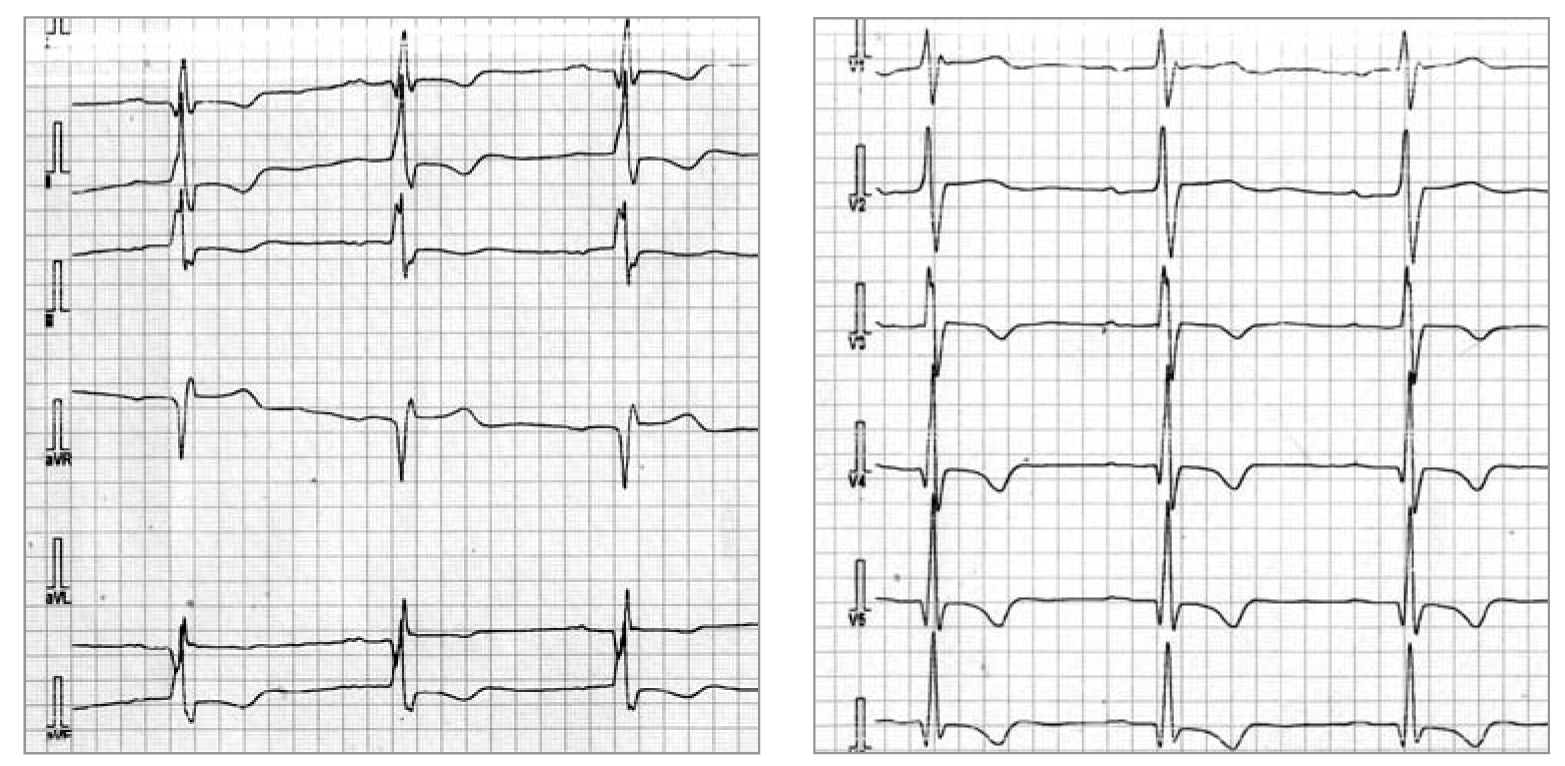 Рис. 2. ЕКГ пацієнтки із хворобою Фабрі. Ритм синусовий, ЧСС 62 уд./хв. AV-блокада I ст. (PQ – 0,22 с), комплекс QRS 0,1 c. Ознаки ГЛШ з його систолічною перегрузкою (негативні Т у відведеннях II, III, AVF, V3-V6; qRs в I, V4-V6; Rsr в V1)
Рис. 2. ЕКГ пацієнтки із хворобою Фабрі. Ритм синусовий, ЧСС 62 уд./хв. AV-блокада I ст. (PQ – 0,22 с), комплекс QRS 0,1 c. Ознаки ГЛШ з його систолічною перегрузкою (негативні Т у відведеннях II, III, AVF, V3-V6; qRs в I, V4-V6; Rsr в V1)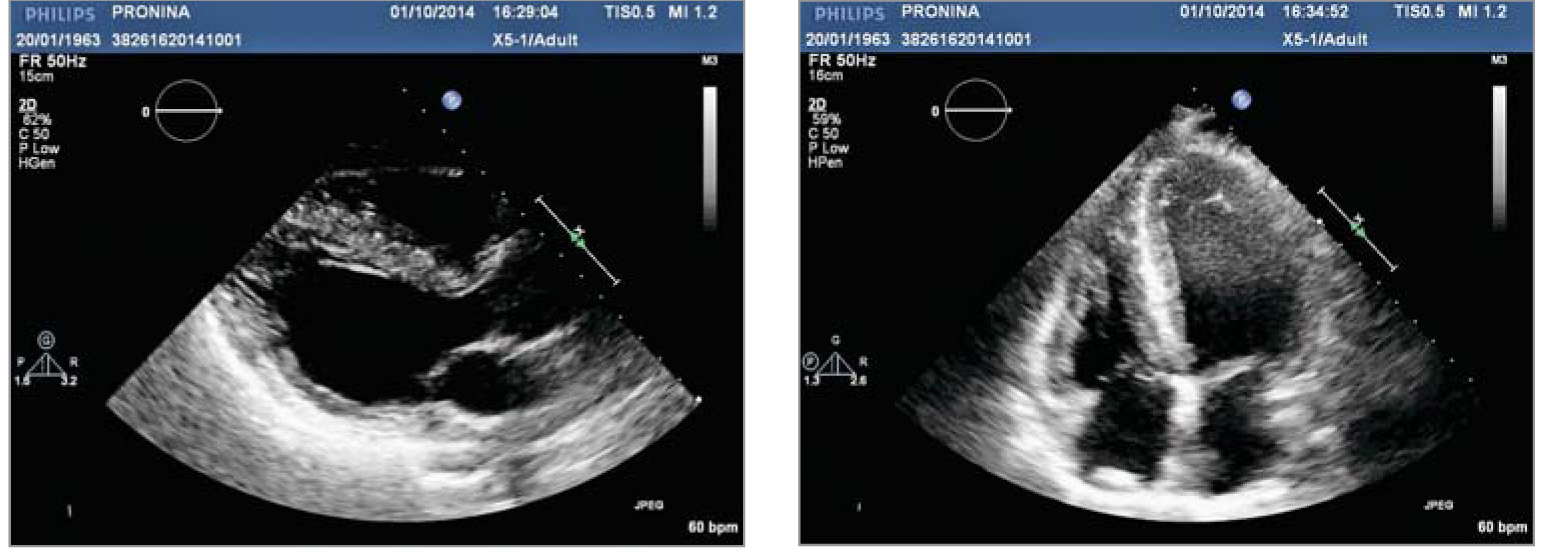 Рис. 3. Трансторакальна ЕхоКГ у парастернальній позиції по довгій осі ЛШ (А) та у верхівковій 4-камерній позиції по довгій осі ЛШ (Б). Відмічаються виражена ГЛШ (переважно за рахунок потовщення МШП), гіпертрофія стінки ПШ, підвищення ехогенності міокарда ЛШ
Рис. 3. Трансторакальна ЕхоКГ у парастернальній позиції по довгій осі ЛШ (А) та у верхівковій 4-камерній позиції по довгій осі ЛШ (Б). Відмічаються виражена ГЛШ (переважно за рахунок потовщення МШП), гіпертрофія стінки ПШ, підвищення ехогенності міокарда ЛШ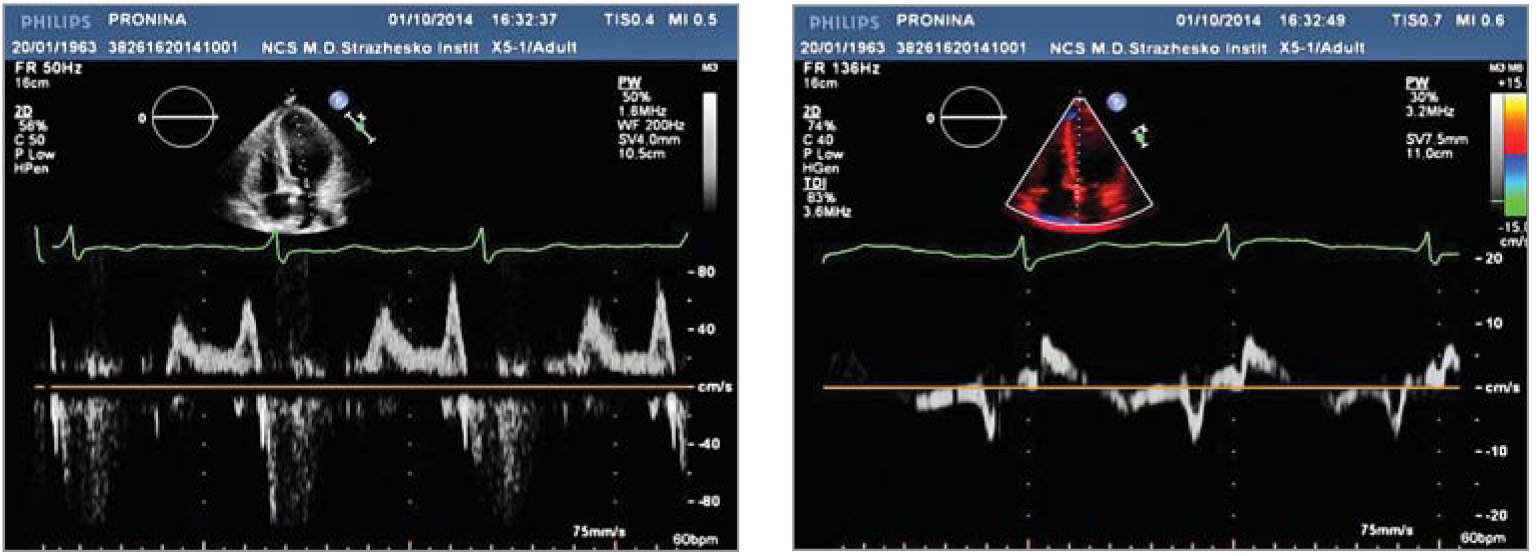 Рис. 4. Показники діастолічної функції ЛШ, отримані за допомогою імпульсно-хвильового допплера (зліва) та тканинного допплера (справа) (діастолічна дисфункція ЛШ 1 типу – порушення релаксації: Е/А=0,7, DTE=230 мс, E/E’=10)
Рис. 4. Показники діастолічної функції ЛШ, отримані за допомогою імпульсно-хвильового допплера (зліва) та тканинного допплера (справа) (діастолічна дисфункція ЛШ 1 типу – порушення релаксації: Е/А=0,7, DTE=230 мс, E/E’=10)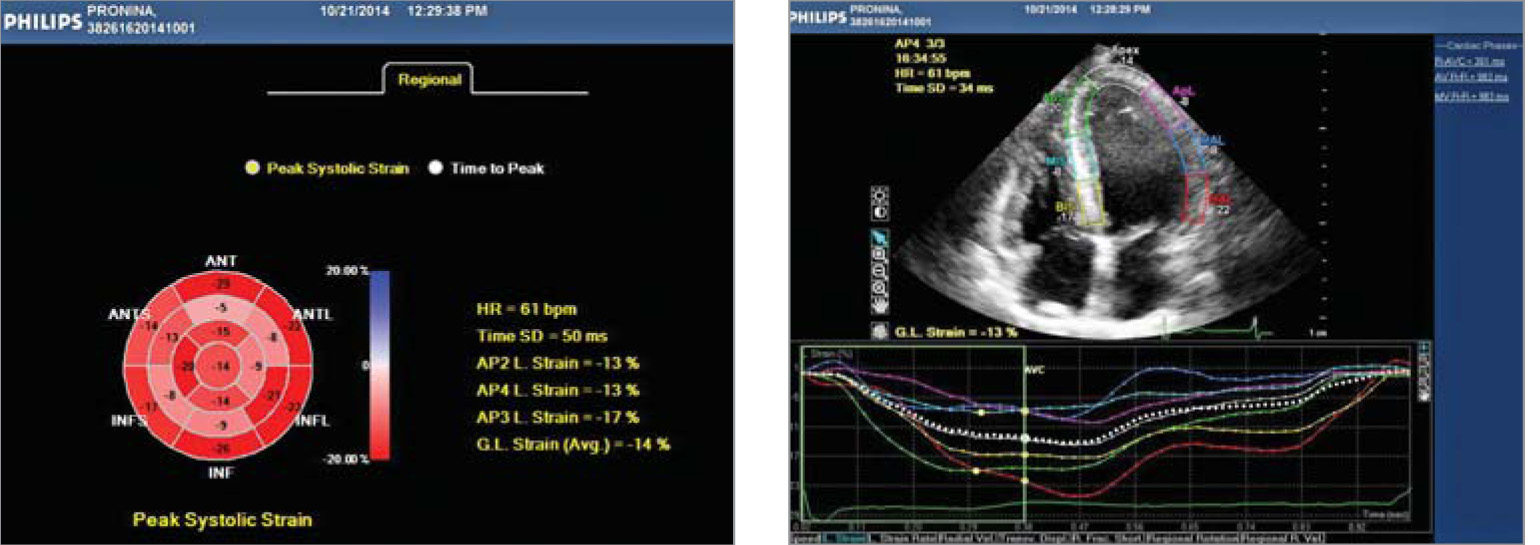 Рис. 5. Двовимірна спекл-трекінг ЕхоКГ виявила зниження глобального поздовжнього стрейну до -14% (в основному за рахунок зниження стрейну середніх та верхівкових сегментів ЛШ), як показано на схематичному зображенні ЛШ у форматі «бичого ока» (А) та на верхівковій 4-камерній позиції ЛШ (Б)
Рис. 5. Двовимірна спекл-трекінг ЕхоКГ виявила зниження глобального поздовжнього стрейну до -14% (в основному за рахунок зниження стрейну середніх та верхівкових сегментів ЛШ), як показано на схематичному зображенні ЛШ у форматі «бичого ока» (А) та на верхівковій 4-камерній позиції ЛШ (Б)Такі зміни на ЕКГ характерні для хвороби Фабрі із ураженням серця.
У хворої не спостерігалося суттєвих порушень ритму за даними добового моніторингу ЕКГ. Було виявлено 39 поодиноких шлуночкових та 41 передсердна екстрасистола. Протягом усього періоду моніторингу виявлялася AV-блокада I ст. із нечастими епізодами AV-блокади II ст., Мобіц I із паузами до 2,4 с (усього 8 пауз у нічний час). Середня ЧСС за добу становила 62 уд./хв із максимальним підвищенням до 98 уд./хв та максимальним зниженням у нічний час до 45 уд./хв.
При проведенні ЕхоКГ була виявлена виражена ГЛШ із асиметричним потовщенням стінок до 2,1 см у середніх сегментах МШП та до 1,3 см вільної стінки ЛШ без ознак значної обструкції виносного тракту ЛШ. Також спостерігалося потовщення стінки ПШ до 1 см (рис. 3). Індекс маси міокарда ЛШ за даними двовимірної ЕхоКГ становив 166 г/м2, що майже в 2 рази перевищує верхню межу норми (до 95 г/м2 для жінок). Виявлено наявність гіперехогенного сигналу, особливо у ділянці МШП та задньої стінки ЛШ, що є ознакою фіброзу і характерно для гіпертрофії, пов’язаної із хворобами накопичення. Фіброз у задньолатеральній стінці ЛШ, що виявляється при проведенні ЕхоКГ та підтверджується при контрастному МРТ-дослідженні серця, свідчить на користь хвороби Фабрі.
Систолічна функція ЛШ збережена, структура та функція клапанів серця не порушені, окрім незначної мітральної регургітації за рахунок потовщення стулок мітрального клапана. Відмічалася незначна динамічна обструкція виносного тракту ЛШ зі збільшенням градієнта тиску з 12 до 23 мм рт. ст.
Також визначалася діастолічна дисфункція ЛШ 1 типу, Е/А=0,7, DTE=230 мс, E/E’=10 (рис. 4), що характерно для ГЛШ. На більш пізніх стадіях ураження серця при хворобі Фабрі можливий розвиток рестриктивного типу діастолічної дисфункції ЛШ.
Систолічна функція ЛШ та ПШ за даними двовимірної ЕхоКГ була збережена, фракція викиду ЛШ становила 65%. Однак при аналізі поздовжньої деформації міокарда ЛШ із використанням двовимірної спекл-трекінг ЕхоКГ було виявлено зниження поздовжнього глобальної деформації (GLS) та швидкості деформації (GLSR), характерне для ГКМП (рис. 5).
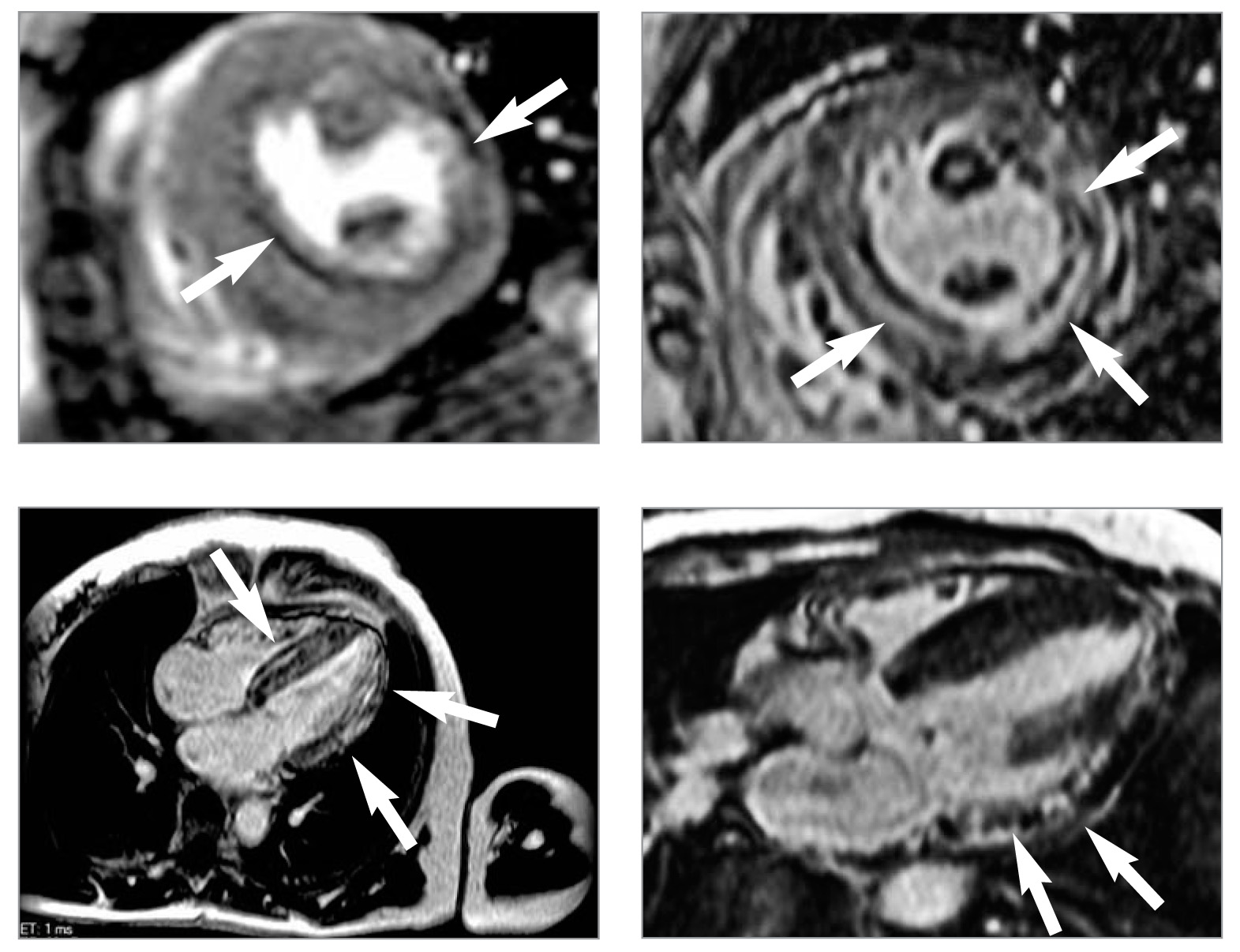 Рис. 6. При проведенні МРТ серця у режимі перфузії (А) спостерігається дефект перфузії міокарда, у режимі пізнього контрастування (Б, В і Г) відмічаються ділянки затримки виведення контрастної речовини, що є ознакою фіброзу (вказано стрілкою). Проекції: А, Б – коротка вісь ЛШ на рівні папілярних м’язів; В – довга вісь ЛШ, 4-камерна проекція; Г – довга вісь ЛШ, 3-камерна проекція
Рис. 6. При проведенні МРТ серця у режимі перфузії (А) спостерігається дефект перфузії міокарда, у режимі пізнього контрастування (Б, В і Г) відмічаються ділянки затримки виведення контрастної речовини, що є ознакою фіброзу (вказано стрілкою). Проекції: А, Б – коротка вісь ЛШ на рівні папілярних м’язів; В – довга вісь ЛШ, 4-камерна проекція; Г – довга вісь ЛШ, 3-камерна проекціяЗ метою оцінки анатомії серця, його структурних змін та можливих порушень перфузії міокарда пацієнтці була проведена МРТ серця із контрастуванням гадолінієм.
У результаті підтверджена гіпертрофія міокарда із потовщенням бокових стінок до 15-16 мм, МШП до 21 мм та масою міокарда 175 г/м2. При контрастуванні були виявлені ознаки фіброзу у боковій та задній стінках ЛШ та МШП (рис. 6): ділянки затримки виведення контрастної речовини (на 12-й та 20-й хвилині) у вигляді інтенсивного магнітно-резонансного сигналу інтрамурально та субендокардіально дифузного характеру в МШП від 2 до 4 мм лінійної форми, у боковій та задній стінках ЛШ до 7-9 мм та частково у верхівці до 3 мм.
Пацієнтка П. могла бути виписана із підтвердженням діагнозу ГКМП, якби пізніше (під час проведення ЕхоКГ та після озвученої підозри на наявність хвороби накопичення) вона не згадала про діагностовану хворобу Фабрі. Жінка не акцентувала увагу на цьому факті з невідомих нам причин. (Ймовірно, вона деякою мірою звинувачує лікарів у пізньому виявленні хвороби як у неї, так і в сина. Хоча порушення у вигляді пекучого болю в стопах ніг, що минув самостійно (без лікування), з’явились у неї в юності (у 15-17 років), а у сина нестерпний біль аналогічної локалізації, який заважав нормальному життю, посилювався під час ходьби та обмежував рухи дитини, виник у віці 5-6 років, лише через 10 років (у віці 16 років) він був обстежений у центрі метаболічних хвороб на базі НДСЛ «Охматдит», де спочатку був запідозрений, а потім підтверджений діагноз хвороби Фабрі.)
Пацієнтка була виписана із діагнозом «Хвороба Фабрі. ГКМП. AV-блокада I ст. Синусова брадикардія. Артеріальна гіпертензія. Серцева недостатність I ст.». Призначено лікування: інгібітори ангіотензинперетворюючого ферменту та дигідропіридиновий блокатор кальцієвих каналів.
Висновки
Діагностика та лікування пацієнтів із хворобою Фабрі потребує участі фахівців різних спеціальностей – педіатрів, нефрологів, неврологів, кардіологів, дерматологів, офтальмологів та ін.; у разі виявлення цієї рідкісної патології необхідно провести обстеження родичів хворого; це особливо важливо для дітей та осіб молодого віку, в яких ферментозамісна терапія може значно покращити прогноз та якість життя. Захворювання зустрічається у гемізиготних чоловіків та гомозиготних або гетерозиготних жінок. У чоловіків зазвичай хвороба має більш тяжкий перебіг із ранніми проявами, для жінок характерні безсимптомний або малосимптомний перебіг, більш пізня маніфестація та повільне прогресування хвороби. Хвороба Фабрі може мати генералізований характер або проявлятися ураженням окремих органів та систем. У практиці кардіолога найбільша вірогідність виявити хворобу у пацієнтів із ознаками ГКМП. Особливістю наведеного клінічного випадку є те, що у жінки середнього віку клінічні прояви хвороби Фабрі були пов’язані з патологією серцево-судинної системи і виявлені за допомогою новітніх методів візуалізації міокарда: ЕхоКГ і МРТ із контрастуванням.
У встановленні діагнозу також можуть допомогти ретельний збір анамнезу і встановлення сімейного характеру захворювання.
* Чоловіки успадковують тільки одну X-хромосому, вони є гемізиготними за всіма генами Х-хромосоми. У разі спадкової передачі Х-зчепленого мутантного гена розвиваються фенотипові прояви захворювання, оскільки Y-хромосома не містить нормальної алелі, що здатна компенсувати функцію мутантного гена.