


11 лютого, 2015
Сучасні засади діагностики та лікування системної склеродермії
А.С. Свінціцький, д.м.н., професор, завідувач кафедри внутрішньої медицини № 3 Національного медичного університету ім. О.О. Богомольця, м. Київ
 Системна склеродермія (ССД) – прогресивне системне захворювання, в основі якого лежить ураження сполучної тканини з переважанням фіброзу, поширена судинна патологія за типом облітераційного ендартерiїту та синдрому Рейно (СР) з характерними фіброзними змінами шкіри, опорно-рухового апарату та внутрішніх органів (легень, серця, травного тракту, нирок).
Системна склеродермія (ССД) – прогресивне системне захворювання, в основі якого лежить ураження сполучної тканини з переважанням фіброзу, поширена судинна патологія за типом облітераційного ендартерiїту та синдрому Рейно (СР) з характерними фіброзними змінами шкіри, опорно-рухового апарату та внутрішніх органів (легень, серця, травного тракту, нирок).
Це захворювання, що проявлялося потовщенням шкіри, вперше згадується в роботах Гіппократа. Термін «склеродермія» («твердошкір’я») ввів у науковий обіг Gintrac у 1847 р., але перший детальний опис хвороби здійснив Zacutus Lusitanus у 1643 р. У подальшому A.G. Maurice Raynaud (1862) вивчав явище «ствердіння шкіри кінцівок», що супроводжувалося зміною забарвлення (побілінням, посинінням, почервонінням). Згодом цей феномен було названо його ім’ям (синдром Рейно), а в 40-х роках XX століття, коли у зв’язку з появою концепції «колагенових хвороб» Клемперера розпочалось інтенсивне вивчення вісцеральної патології при склеродермії, було описано її системний характер і склеродермічну групу захворювань (СГЗ).
ССД належить до СГЗ, що включає також обмежену (вогнищеву) склеродермію, дифузний еозинофільний фасцiїт, склеродерму Бушке, мультифокальний фіброз, індуковані форми склеродермії та псевдосклеродермічні синдроми. Goetz запропонував термін «прогресуючий системний склероз» для визначення склеродермії як системного захворювання.
Епідеміологія. ССД за частотою посідає третє місце серед усіх системних захворювань сполучної тканини (СЗСТ). За останні десятиріччя спостерігається зростання захворюваності на ССД до 5-16 випадків на 1 млн населення на рік. ССД дебютує переважно у віці 30-50 років. Жінки хворіють у 3-5 разів частіше, ніж чоловіки, і найбільш часто – у дітородному віці.
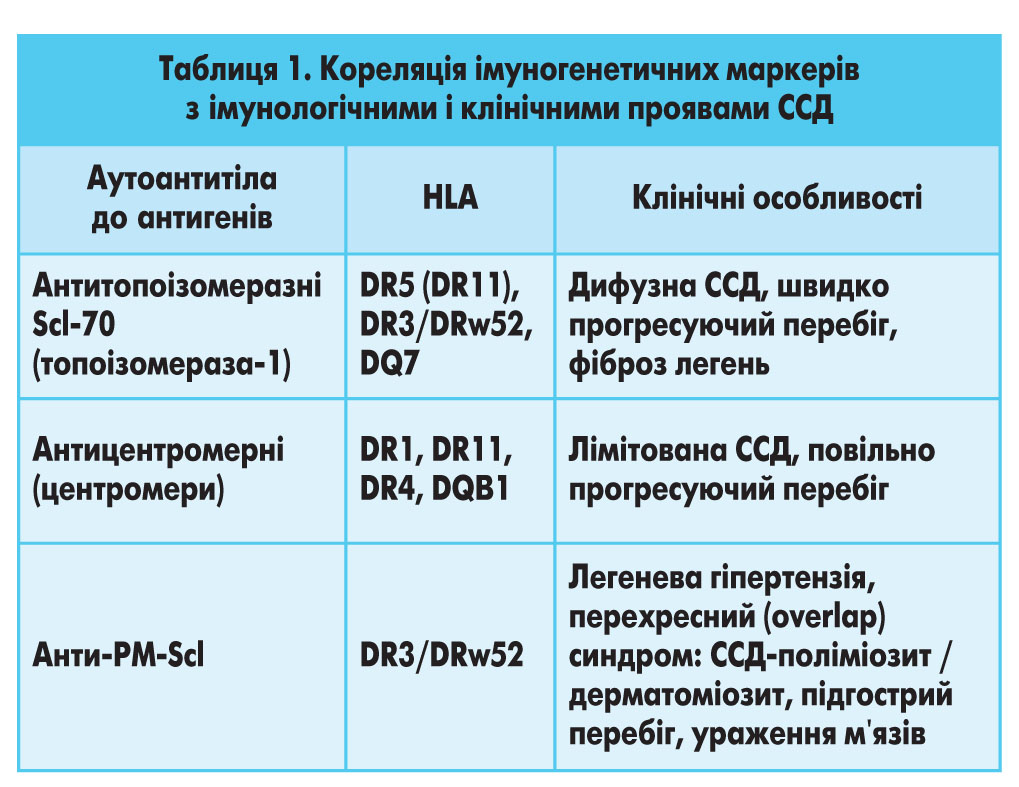
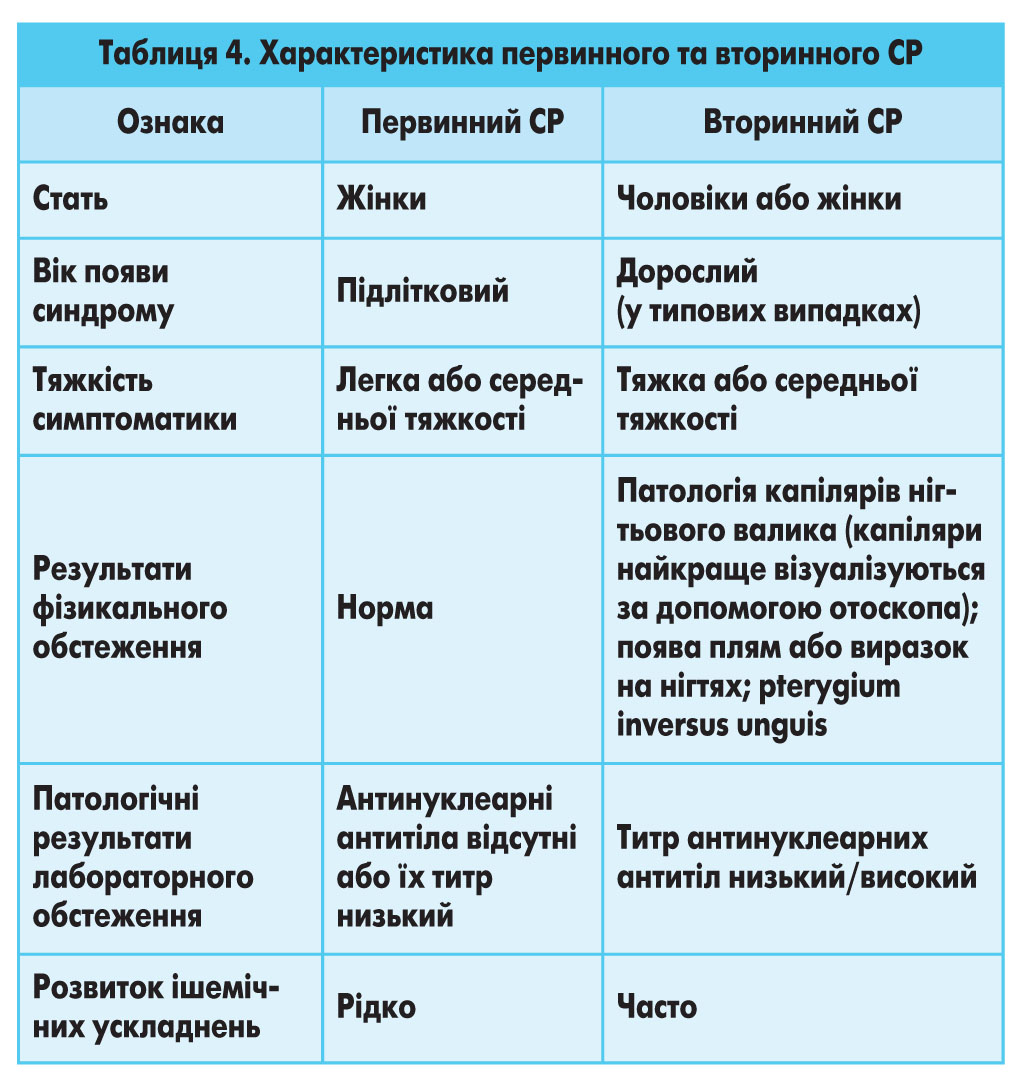
Етіологія. ССД розглядають як взаємодію несприятливих екзо- й ендогенних чинників за наявності генетичної схильності до захворювання. У пацієнтів із ССД підтверджено існування хромосомної нестабільності. Наявність сімейних випадків ССД і близьких захворювань (системний червоний вовчак – СЧВ, ревматоїдний артрит – РА, синдром Шегрена, СР і його еквівалент, кардіо- і нефропатії нез’ясованого ґенезу, ураження щитоподібної залози тощо), частину з яких можна розглядати як неповний прояв ССД, виявлення імунологічних та інших лабораторних порушень у здорових родичів пробандів, висока частота хромосомних аномалій і асоціація ССД з певними антигенами системи гістосумісності НLА А9, В8, В35 DR3, DR5, DR11, DR52 та Сw4, відповідальними за імунну відповідь, підтверджують участь генетичних механізмів у розвитку зазначеного захворювання.
Принципово важливою є кореляція імуногенетичних маркерів з імунологічними (специфічні антитіла) і клінічними проявами ССД (табл. 1).
Очевидним ендогенним фактором ризику розвитку ССД є гормональний дисбаланс, про що свідчить виражений статевий диморфізм.
Поряд з інфекцією (ретро-, герпесвіруси тощо), вакцинацією, переохолодженням, вібрацією, травмами (зокрема, хірургічними втручаннями), стресами й ендокринними змінами (вагітність, пологи, аборт, менопауза) тригерний ефект зумовлюють хімічні агенти (промислові, побутові, аліментарні, медикаментозні).
Існує взаємозв’язок ССД і злоякісних пухлин; крім розвитку різних варіантів псевдосклеродермічного паранеопластичного синдрому при злоякісних новоутвореннях можливим є також виникнення в онкологічних хворих типової форми ССД.
Патогенез. Основні патогенетичні механізми ССД (рис. 1):
· патологічне фіброзоутворення;
· імунні розлади;
· порушення мікроциркуляції.
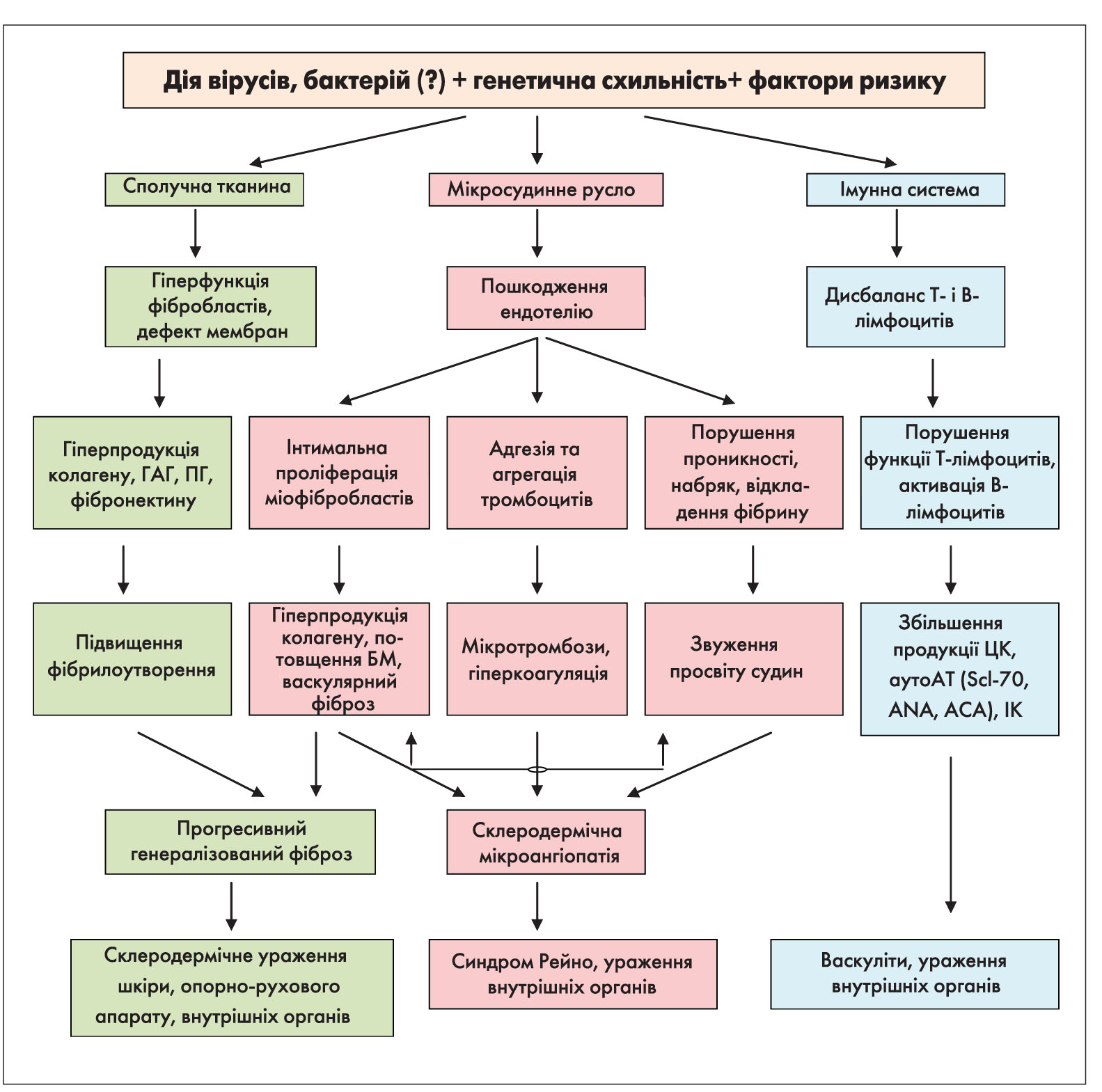 Рис. 1. Патогенез ССД
Рис. 1. Патогенез ССДЦентральне місце в патогенезі ССД належить процесам посиленого колагено- і фіброзоутворення. Доведено, що властиві захворюванню індуративні зміни шкіри (так само, як і фіброз вісцеральних органів) зумовлені значним посиленням біосинтезу колагену фібробластами, подальшим збільшеним неофібрилогенезом. Більше того, виявлено надмірну продукцію фібронектину і глікопротеїну. Активацію фібробластів у хворих на ССД підтверджено шляхом гібридизації клонів cDNA in situ.
Фенотипічно стійка гіперфункція склеродермічних фібробластів із посиленням біосинтезу колагену та неофібрилогенезу, збільшення вмісту фібронектину та інших компонентів матриксу, функціональна та структурна дефектність клітинних мембран і рецепторів спричиняють відносну автономність фібробластів при ССД. Припускають первинний чи індукований метаболічний дефект фібробластів, який реалізується під впливом екзогенних чинників (мутантний ген фібриліну); порушення апоптозу при ССД є можливим чинником селекції фенотипу «склеродермічних» фібробластів.
Носійство певних НLА-антигенів зумовлює схильність до продукції різних аутоантитіл (аутоАТ) та формування відповідних клінічних синдромів і субтипів захворювання. При ССД виявлено тісний взаємозв’язок та участь у розвитку склеродермічного процесу принаймні 6 основних видів клітин:
1) фібробласти;
2) ендотеліоцити;
3) гладком’язові – здатні до вазоконстрикції та фібробластної активності;
4) імунокомпетентні – Т-лімфоцити та моноцити;
5) опасисті клітини, які активують ендотелій та фібробласти;
6) тромбоцити – джерело факторів росту та інших цитокінів.
Міжклітинна кооперація здійснюється за допомогою різних медіаторних систем: молекул адгезії, факторів росту – трансформуючих, тромбоцитарних і фібробластних (TGFβ, PDGF, FGFβ), фактора некрозу пухлини (TNF), ендотеліну-1 та інших вазоактивних пептидів, інтерферонів та інтерлейкінів (1, 2, 4, 6, 8), частина з яких потенційно мають фіброгенний ефект.
Важливою ланкою патогенезу ССД є мікроциркуляторні порушення, ураження судинної стінки, зміни реологічних властивостей крові (збільшення агрегації еритроцитів, тромбоцитів, вазоконстрикція, мікротромбоз) з розвитком СР. При цьому першою уражається базальна мембрана судин та ендотелій, далі спостерігається потовщення інтими, що супроводжується зменшенням внутрішнього діаметра судин, їх облітерацією, підвищеною чутливістю і здатністю до вазоконстрикції малих судин. У процесі облітерації малих судин посилюється стан ішемії шкіри та внутрішніх органів. Окрім облітерації судин, у розвитку ішемії органів певну роль відіграє судинний спазм, у формуванні якого беруть участь вазоактивні субстанції (ендотелін-1), а також дефіцит судинорозширювальних нейропептидів, активація тромбоцитів із збільшенням виділення тромбоцитарного фактора росту та подальшою проліферацією фібробластів і синтезом колагену.
На фоні ураження стінки судин спостерігаються зміни внутрішньосудинних плазменних і клітинних властивостей крові: підвищення в’язкості крові, тенденція до гіперкоагуляції, пригнічення фібринолізу та агрегації формених елементів, підвищення вмісту білкових макромолекул і розвиток синдрому дисемінованого внутрішньосудинного згортання крові (ДВЗ-синдрому), що супроводжується ішемічними та некротичними локусними змінами. Ураження ендотелію призводить до активації гіперактивних фібробластів із посиленням біосинтезу колагену та інших компонентів сполучної тканини і розвитком регіонального або генералізованого фіброзу. При ССД ендотеліальна активація супроводжується запальними змінами і фіброзом, що зумовлює прогресування обструктивної васкулопатії і в підсумку – виражену дисфункцію і пошкодження внутрішніх органів.
Важливу роль у прогресуванні аутоімунного процесу при ССД відіграють також аутоАТ IgG-ізотипу, причому цитотоксична активність сироваток щодо молекул- і клітин-мішеней сполучної тканини визначається не лише афінністю та ідіотипічним спектром аутоАТ, а й співвідношенням у популяції таких аутоАТ високо- і низькоавідних фракцій, коли високоавідні аутоАТ IgG-ізотипу полівалентно зв’язуються з епітопами антигену і призводять до реалізації ефекторних функцій АТ.
Класифікація. Асоціацією ревматологів України (2004) до використання рекомендовано класифікацію ССД, наведену в таблиці 2.
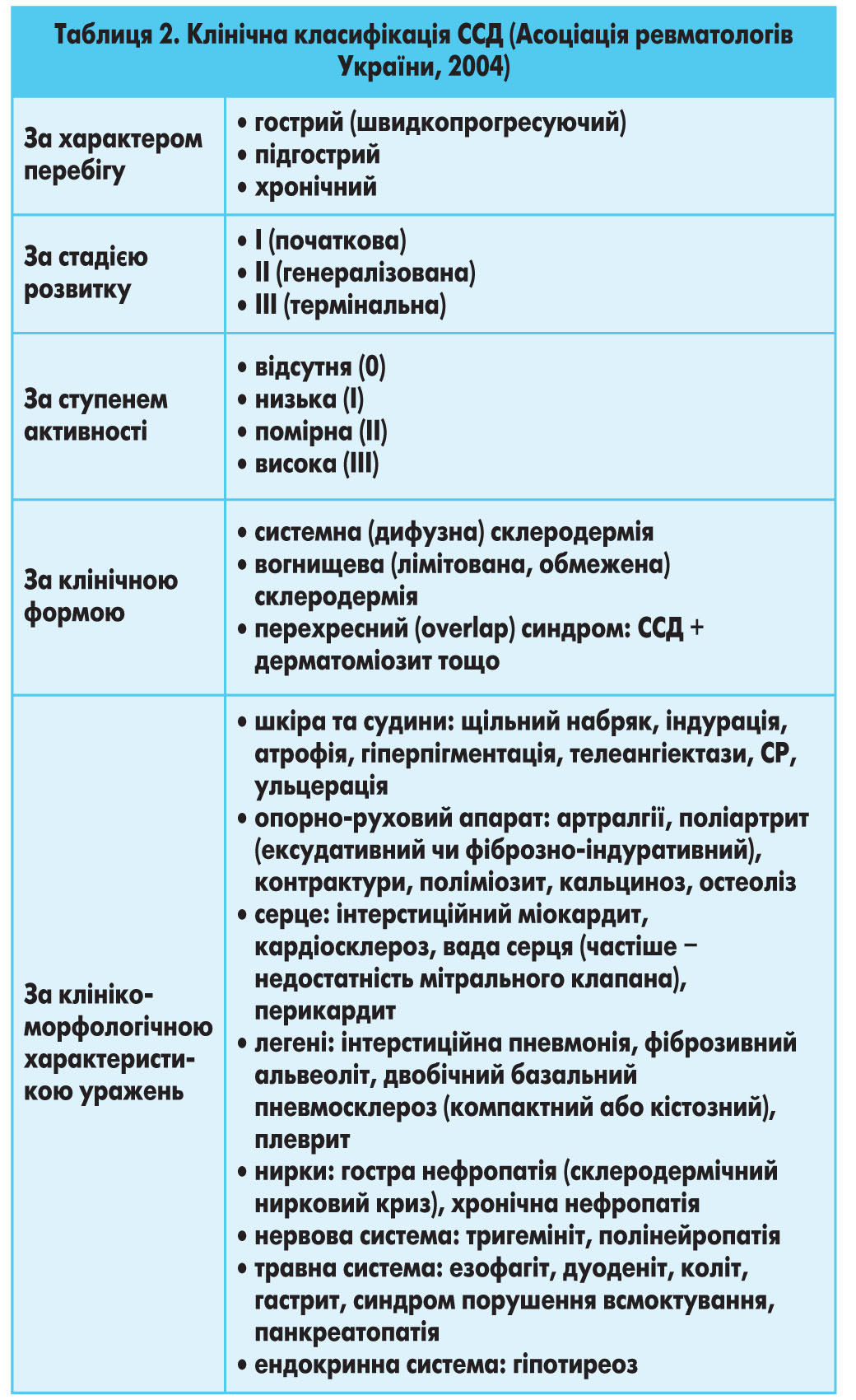
За наявності ураження серця, легень, нирок та опорно-рухового апарату рекомендується вказувати функціональну недостатність (серцева, дихальна, ниркова, суглобів тощо) відповідно до існуючих класифікацій.
Провідні світові експерти пропонують виділяти у клінічній практиці такі форми ССД:
1) обмежена (лімітована) форма:
• розвивається поступово, прогресує повільно, але постійно; ураження шкіри обмежене ділянкою обличчя та кистей/стоп;
• першим проявом зазвичай є СР, який може випереджати появу інших симптомів хвороби на 3-12 років;
• судинні симптоми (СР, виразкування пальців, телеангіектазії) часто домінують у клінічній картині від самого початку хвороби, a органні ускладнення (порушення ковтання, інтерстиційна хвороба легень, легенева гіпертензія) з’являються пізніше;
• одним із варіантів є СRЕSТ-синдром: підшкірний кальциноз (calcinosis), феномен Рейно (Reynaud phenomenon), порушення моторики стравоходу (esophageal motility disorders), склеродактилія (sclerodactyly) і телеангіектазії (teleangiectasis); виявляються антицентромерні антитіла;
• спостерігаються підвищені рівні прозапальних цитокінів, антитіл до ДНК, стимуляція фібробластів; велике значення в регуляції профіброзних аспектів вродженої імунної активації відіграють трансформуючий фактор росту β (TGFβ) у присутності епідермального фактора росту (EGF);
• при вільнорадикальному окислюванні нейтрофілів пацієнти з локалізованою склеродермією мають дисфункції кількості лімфоцитів CD3+, CD4+, CD11b+ при підвищеній кількості лімфоцитів CD8+, CD16+, а також маркерів активації CD19+, CD25+, CD95+ і HLA-DR+-клітин;
• прогноз відносно сприятливий: 10-річна виживаність становить понад 80%;
2) дифузна форма:
• початок хвороби зазвичай раптовий, характеризується симетричним потовщенням шкіри обличчя, тулуба, кінцівок протягом року, а також швидким залученням до патологічного процесу внутрішніх органів (інтерстиційне ураження легенів) і подальшим швидким прогресуванням;
• шкірні зміни, судинні і суглобові симптоми з’являються майже одночасно або з невеликим відривом у часі (зазвичай до року);
• реєструють ураження шлунково-кишкового тракту (ШКТ), міокарда, нирок;
• спостерігається значна редукція капілярів нігтьового ложа з формуванням аваскулярних ділянок (за даними капіляроскопії нігтьового ложа); виявляються антитіла до топоізомерази-1 (Scl-70);
• тяжкі органні ускладнення (інтерстиційна хвороба легень, склеродермічний нирковий криз, ураження міокарда, ускладнення з боку ШКТ) з’являються вже на початку хвороби;
• мoжe виникати тертя сухожилків; антицентромерні антитіла не визначаються;
• через декілька років перебіг хвороби стабілізується, а шкіра може поступово стоншуватися;
• прогноз є серйозним: 10-річна виживаність становить 40-60%.
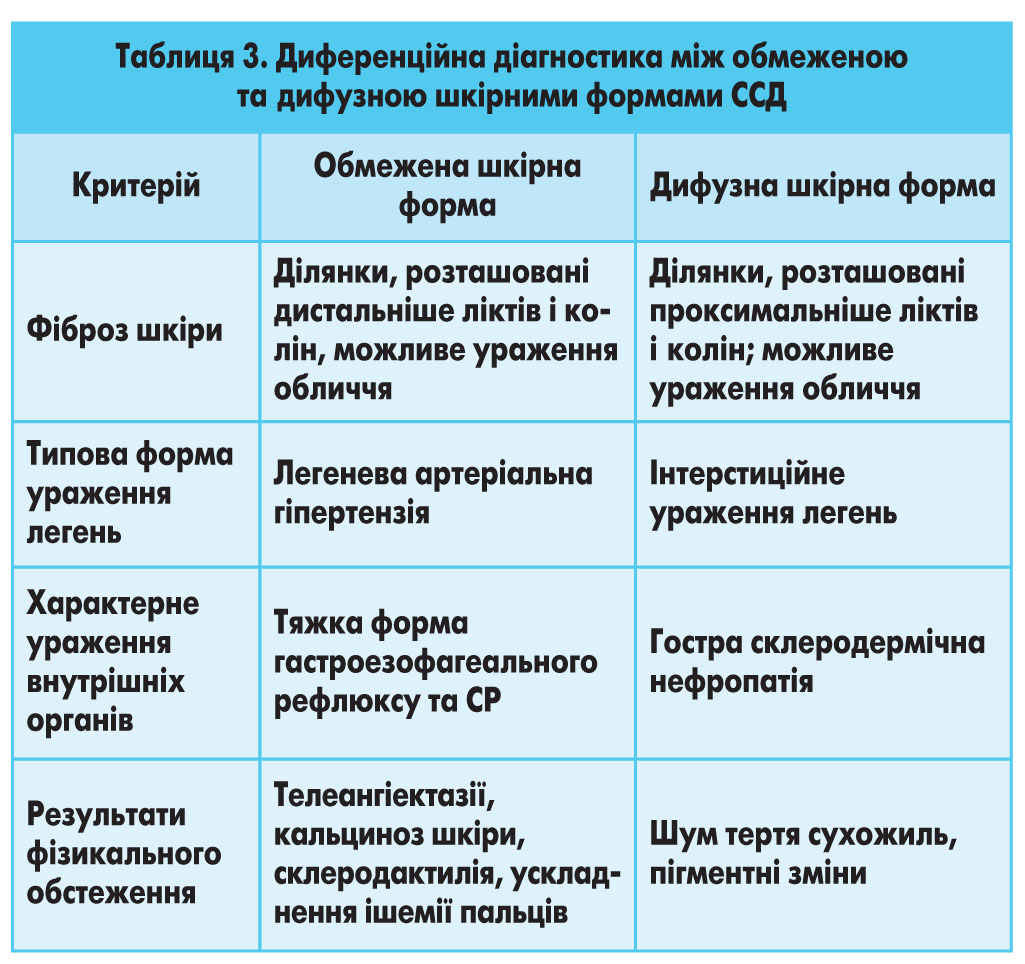
Відмінності обмеженої та дифузної шкірних форм ССД представлено в таблиці 3;
3) ССД без шкірних змін (вісцеральна форма):
• типові прояви з боку внутрішніх органів і систем із супутніми характерними органними змінами (виявляються феномен Рейно, ознаки легеневого фіброзу, гострої склеродермічної нирки, ураження серця та ШКТ);
• наявність змін серологічних показників, властивих для ССД: антинуклеарні антитіла (Scl-70, нуклеолярні);
• відсутність шкірних змін;
4) перехресна форма (оvеrlар sуndrоme) – синдром поєднання клінічних ознак ССД із симптомами іншого СЗСТ (найчастіше РА, дерматоміозит, СЧВ, змішане захворювання сполучної тканини – ЗЗСТ);
5) синдром значного ризику розвитку ССД (пресклеродерма):
• СР;
• ознаки, властиві для ССД при капіляроскопії, та специфічні для ССД АNА (АСА, Scl-70 або протиядерцеві антитіла);
• відсутність затвердіння шкіри й органних змін;
• у близько 65% осіб із цим синдромом протягом 5 років розвивається ССД (переважно обмежена форма).
Клінічна картина. Ураження шкіри є основним симптомом, проте спостерігається не у всіх пацієнтів із ССД, особливо на початку захворювання. Шкірні зміни при ССД проходять три стадії: щільного набряку, індурації та атрофії. Під час І стадії виявляють набряк шкіри, почервоніння або блідість. Ця стадія незабаром переходить у другу, найбільш тривалу – стадію індурації або фіброзу: шкіра стає сухою, гладкою, спаяною з прилеглими тканинами, іноді спостерігається її пігментація або кератоз; стає чітким судинний малюнок, з’являються телеангіектази, переважно на обличчі та грудях. Третя стадія – стадія атрофії – характеризується значним стоншенням шкіри, пігментацією, водночас спостерігається атрофія прилеглих тканин.
Шкіра обличчя стає натягнутою, обличчя – маскоподібним, амімічним, ніс загострюється («пташиний дзьоб»), ніздрі натягуються, губи стоншуються, шкіра навколо них збирається в складки, які спрямовані до ротової щілини («кисетний шов»). З часом хворий втрачає здатність широко відкривати рот, що може залежати і від ураження скронево-нижньощелепних суглобів. Характерні трофічні порушення (алопеція, оніходистрофія, трофічні виразки та рубчики, ангідроз, гіперкератоз шкіри). Поступове ураження шиї та грудної клітки викликає у хворого відчуття «панцира», виникає поверхневе дихання та задишка. При дифузній формі ССД ураження шкіри на різних ділянках інколи бувають різного ступеня вираження та стадії патологічного процесу, тотальне залучення шкіри тулуба й кінцівок призводить до муміфікації та кахексії («живі мощі») (рис. 2). Ураження шкіри (шкірний склероз) є одним із перших клінічних проявів хвороби, що спричиняє страждання хворого. Приєднання вісцеральної патології суттєво погіршує прогноз і зумовлює значне зниження якості життя у цієї категорії пацієнтів.
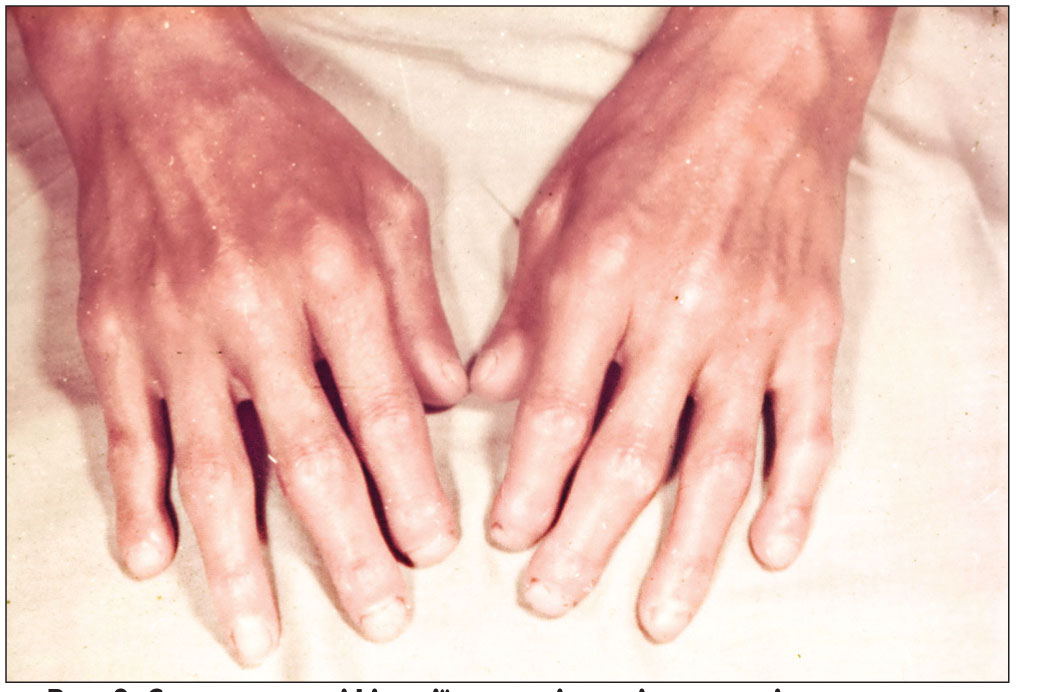 Рис. 2. Симптом муміфікації – «живі мощі», остеоліз дистальних фаланг при ССД
Рис. 2. Симптом муміфікації – «живі мощі», остеоліз дистальних фаланг при ССДЧасто уражаються слизові оболонки (хронічний кон’юнктивіт, атрофічний риніт, фарингіт, стоматит). У 5-20% хворих виникає синдром Шегрена, який включає:
• лімфаденопатію – регіонарну (збільшення підщелепних, шийних, надключичних лімфовузлів) або генералізовану;
• збільшення привушних слинних залоз, зазвичай – двобічне («мордочка хом’яка»); сухість носової частини глотки, сухі кірки в носі та євстахієвій трубі, сухість голосових зв’язок, осиплість голосу;
• лімфоїдну інфільтрацію паренхіми слинних і сльозових залоз із їх атрофією;
• сухість слизових оболонок порожнини рота (рис. 3), очей з розвитком ксерофтальмії та ксеростомії («сухий катар»);
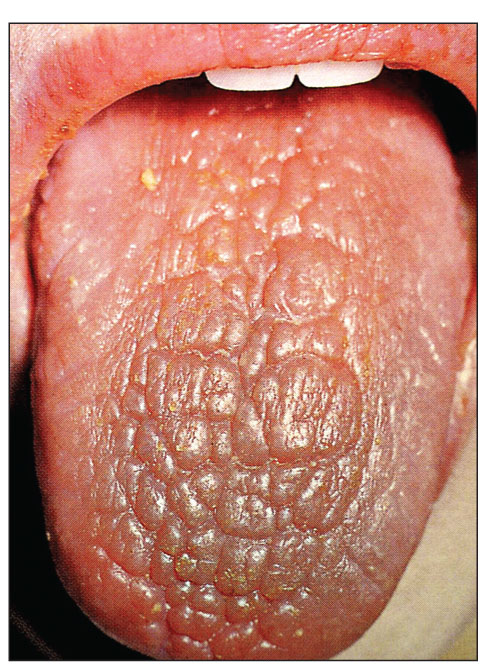 Рис. 3. Синдром Шегрена при ССД
Рис. 3. Синдром Шегрена при ССД• рецидивні артралгії з незначною вранішньою скутістю; у третини хворих – міалгії;
•дисфагію (зумовлену ксеростомією та гіпокінезією), шлункову диспепсію (важкість в епігастрії після їди, відрижка повітрям, нудота, зниження апетиту);
• у 60-70% хворих – розвиток хронічного атрофічного гастриту, ахлоргідрії; виникнення токсичного гепатиту.
Ураження опорно-рухового апарату. Суглобовий синдром може бути ранньою ознакою, розвивається майже у всіх хворих на ССД, буває трьох варіантів: 1) поліартралгії; 2) поліартрит – переважно з ексудативно-проліферативними чи фіброзними змінами; 3) периартрит – здебільшого зі слабко вираженим больовим синдромом, з розвитком контрактур (м’якотканинного походження) та дефігурацій суглобів («пташина лапка», ревматоїдоподібна артропатія за типом Жаку) (рис. 4). Ураження м’язів при ССД проявляється переважно у вигляді інтерстиційного міозиту, що супроводжує фіброзуючі процеси в суглобах і м’яких тканинах (при цьому м’язи ущільнені, напружені, з часом атрофуються), та поліміозиту із первинними запально-дегенеративними та некротичними змінами м’язових волокон, що проявляється міалгіями, м’язовою слабкістю та скутістю, гіперферментемією. Ураження суглобів і м’язів за типом РА та склерозивного міозиту – не лише частий клінічний синдром, а й одна з основних причин ранньої інвалідності хворих на ССД.
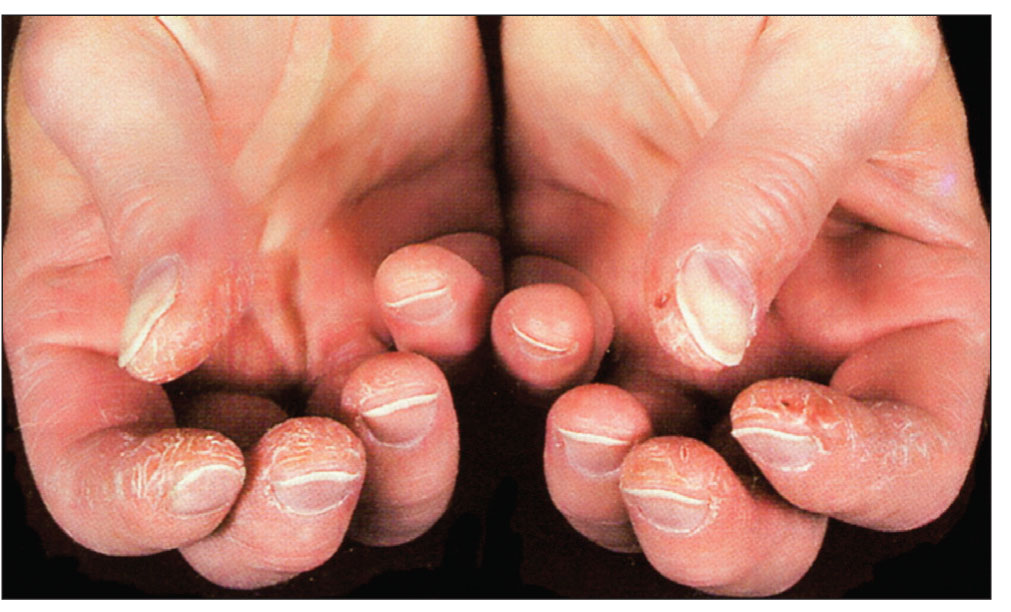 Рис. 4. Ураження кистей при ССД: склеродактилія, симптом «пташині лапки»
Рис. 4. Ураження кистей при ССД: склеродактилія, симптом «пташині лапки»Досить характерною ознакою ССД є остеоліз нігтьових фаланг кистей, унаслідок чого розвиваються деформації та укорочення кистей. При цьому кальцифікація м’яких тканин спостерігається у 30% хворих, переважно в тканинах пальців рук та периартикулярно – навколо ліктьових, плечових, кульшових суглобів, у підшкірній клітковині, інколи за ходом фасцій, сухожиль; поверхневі білі вогнища кальцифікатів просвічуються крізь шкіру (синдром Тіб’єржа-Вайссенбаха) (рис. 5).
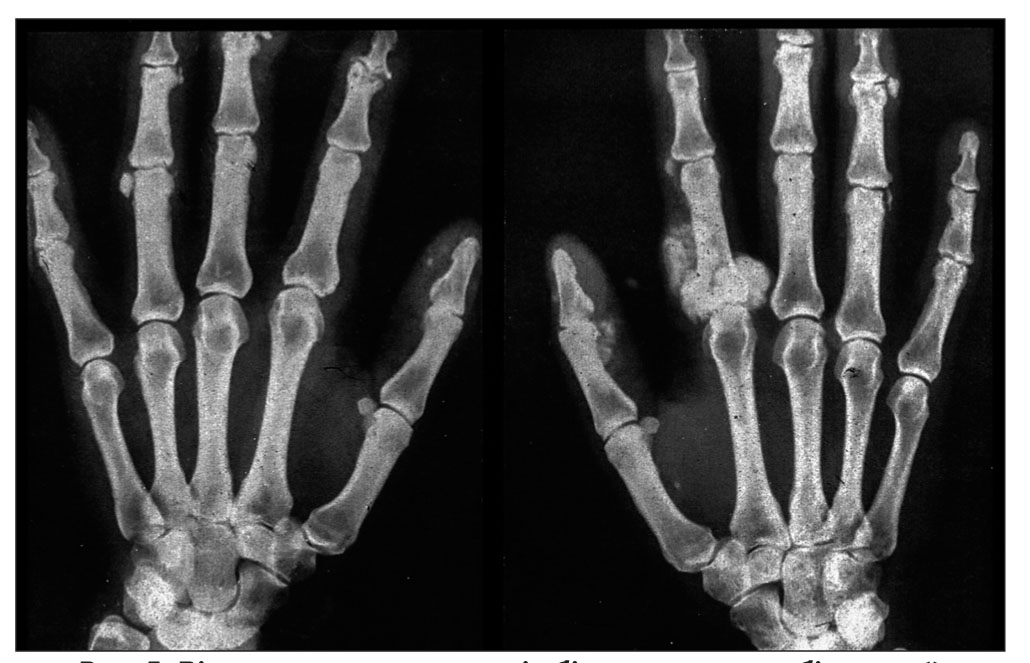 Рис. 5. Відкладення кальцинатів біля малих суглобів кистей (симптом Тіб’єрже-Вайссенбаха)
Рис. 5. Відкладення кальцинатів біля малих суглобів кистей (симптом Тіб’єрже-Вайссенбаха)
Одним із найбільш ранніх і практично постійних проявів ССД є СР. На початку хвороби СР проявляється епізодично, залучаються дистальні відділи кількох пальців рук, а потім і повністю кистей, стоп, рідше – носа, губ, вух. З часом на кистях розвиваються згинальні контрактури, а згодом – склеродактилія, акросклероз, через остеоліз окремих фаланг вкорочуються пальці.
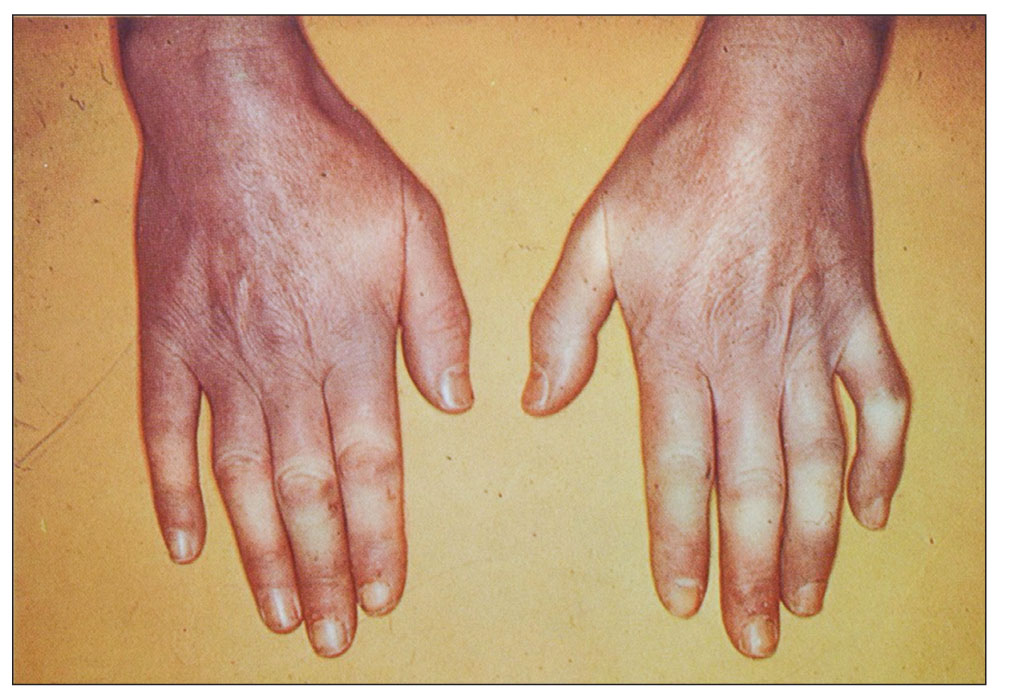 Рис. 6. Кисті хворої на обмежену форму ССД, СР
Рис. 6. Кисті хворої на обмежену форму ССД, СРІдіопатичний, або первинний, СР зазвичай виникає у дівчаток підліткового віку і не супроводжується ішемічними ускладненнями. Вторинний СР, навпаки, розвивається пізніше і часто призводить до ураження тканин. Під час фізикального обстеження при вторинному СР можна виявити ціаноз та ознаки ішемічного ураження пальців (рис. 6), у тому числі появу плям, видимих капілярів на нігтьовому ложі та pterygium inversus unguis (зрощення дистального ложа нігтя з вентральною поверхнею нігтьової пластинки) (табл. 4).
Ураження травної системи виникає у 60-70% пацієнтів із ССД. До патологічного процесу можуть залучатися всі відділи травного тракту, але найчастіше – стравохід і кишечник.
Зміни стравоходу є ранніми ознаками ССД практично у всіх хворих (у 50% з них – безсимптомні). Під час рентгенологічного дослідження виявляють порушення моторики стравоходу, регургітацію їжі у стравохід, рефлюкс-езофагіт. Дисфагія дещо зменшується при ковтанні у вертикальній позиції та посилюється в міру прогресування хвороби. Майже 2/3 пацієнтів висловлюють такі скарги вже на ранньому етапі розвитку хвороби, а в низці випадків вони навіть передують шкірним змінам.
Ураження шлунка супроводжується частим блюванням, тонкої кишки – здуттям, розпиранням, болем, а згодом – порушенням всмоктування, діареєю, зменшенням маси тіла.
У 30% хворих виявляють ураження порожнини рота: атрофію сосочків язика з подальшим порушенням смаку, загрубіння слизової оболонки ямкових відростків, атрофію щільної пластини і запалення ясен, що призводить до втрати зубів.
Кровотечі з уражених судин верхнього відділу травного каналу є основною причиною розвитку анемії у пацієнтів із ССД.
Під час клінічного обстеження найчастіше виявляють помірну гепатомегалію або гепатолієнальний синдром, симптоми первинного біліарного цирозу печінки, іноді – з ознаками портальної гіпертензії. Рідше розвивається фіброз підшлункової залози з недостатністю її секреторної функції.
Ураження серцево-судинної системи є одним з основних вісцеральних проявів. Склеродермічний кардіосклероз клінічно характеризується помірним болем у ділянці серця, задишкою, екстрасистолією, приглушенням тонів і систолічним шумом на верхівці, розширенням серця вліво. Серцева недостатність розвивається рідко. Ураження пристінкового ендокарда спостерігається майже у всіх пацієнтів, інколи виникає фібропластичний ендокардит.
Ураження легень характеризується клініко-рентгенологічною симптоматикою базального, а надалі – і дифузного пневмосклерозу. Особливостями пневмосклерозу при ССД є раннє ураження судин із розвитком симптоматики легеневої гіпертензії та навантаження на праві відділи серця, хронічний сухий кашель. Рентгенологічно виявляють характерне посилення легеневого малюнка, його деформацію. Під час комп’ютерної томографії легень з високою роздільною здатністю реєструють грубу, тяжисту або кістозну перебудову легеневої тканини («стільникові» легені) (рис. 7).
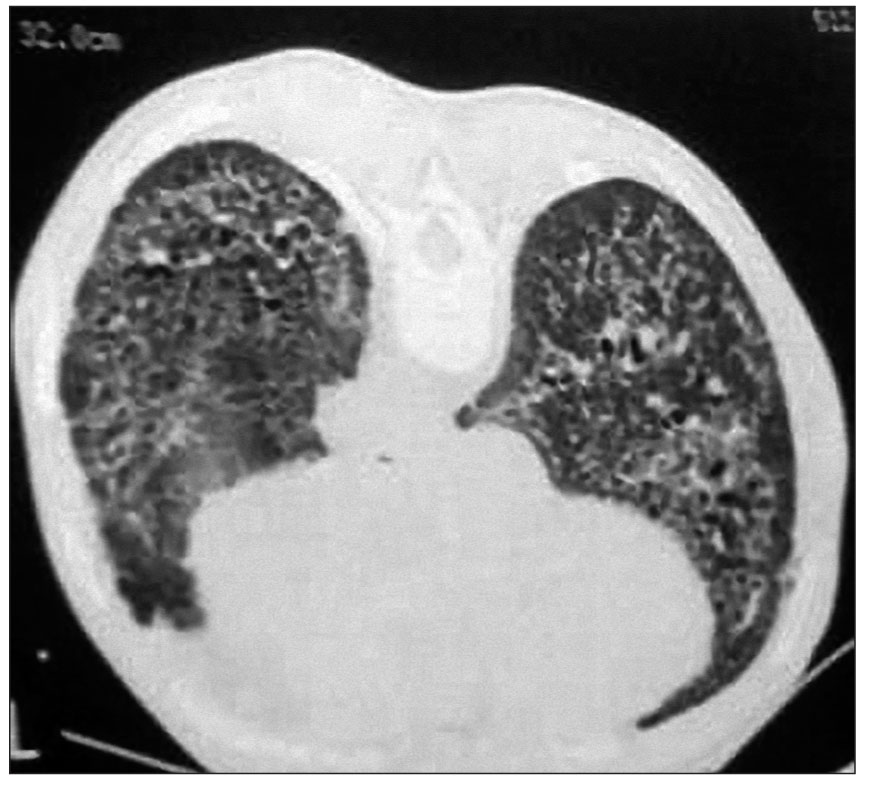 Рис. 7. Двобічний базальний пневмофіброз («кістозні легені» у хворої на ССД)
Рис. 7. Двобічний базальний пневмофіброз («кістозні легені» у хворої на ССД)У третини хворих виникає ураження нирок, переважно у вигляді сечового синдрому (СС). До впровадження у клінічну практику інгібіторів ангіотензинперетворювального ферменту найвищий рівень смертності спостерігався при такому ускладненні ССД, як гостра склеродермічна нефропатія (справжня склеродермічна нирка, склеродермічний нирковий криз). Це ускладнення розвивається у 3-10% усіх хворих із ССД та у 10-20% пацієнтів із швидкопрогресуючою дифузною шкірною формою ССД. При цьому найвищий ризик спостерігається протягом перших трьох років захворювання. До інших факторів ризику належать застосування кортикостероїдів у високих дозах (понад 15 мг на добу), наявність шуму тертя сухожиль, безсимптомного випоту в перикарді, поява анемії, похилий вік та вагітність. При гострій нефропатії у хворих раптово підвищується рівень артеріального тиску, що супроводжується прогресуючою олігуричною нирковою недостатністю з протеїнурією, мікроангіопатичною анемією та мікрогематурією. У 10-15% пацієнтів з гострою нефропатією артеріальний тиск нормальний, але порівняно з початковим рівнем він підвищений. Тому для ранньої діагностики гострої нефропатії дуже важливо регулярно вимірювати рівень артеріального тиску.
У хворих на ССД можливим є розвиток вторинного антифосфоліпідного синдрому, який у разі рецидивуючої втрати вагітності варто верифікувати за наявності відповідних додаткових проявів. З огляду на вищенаведене раціональною є оцінка стану антифосфоліпідних антитіл у пацієнток із ССД перед зачаттям і, безумовно, у разі безпліддя чи невиношування вагітності.
У перебігу ССД виділяють такі варіанти.
Для хронічного (найбільш частого) перебігу ССД притаманні прогресивні вазомоторні порушення у вигляді СР та зумовлені ним трофічні розлади. У подальшому у клінічній картині переважають вазомоторні порушення з поступовим розвитком ущільнення шкіри, утворенням контрактур, остеолізом і повільно прогресуючими склеродермічними змінами внутрішніх органів, переважно стравоходу. П’ятирічна виживаність сягає 88%.
Підгострий перебіг характеризується появою щільного набряку шкіри з подальшою індурацією, рецидивного поліартриту, рідше – міозиту з міастенічним синдромом, полісерозиту, вісцеральної патології (інтерстиційної пневмонії з розвитком пневмосклерозу, міокардиту, склеродермічного езофагіту й ураження всіх інших кишок, патології нирок за типом склеродермічної нирки) на фоні нерізко виражених вазомоторних трофічних порушень. Для 75% хворих виживаність становить 5 років.
Гострий перебіг характеризується тяжкими периферійними та вісцеральними ураженнями з розвитком функціональної недостатності органів уже в перші місяці хвороби, ранньою генералізацією процесу, неухильним прогресуванням і летальним наслідком упродовж двох років; при цьому п’ятирічна виживаність становить лише 4%.
Стадії захворювання:
І – переважно суглобові прояви при підгострому перебігу та вазоспаcтичні – при хронічному;
ІІ – стадія генералізації процесу, характеризується полісиндромністю та полісистемністю ураження багатьох органів і систем;
III – термінальна з переважанням тяжких склеротичних, дистрофічних або судинно-некротичних процесів, нерідко з порушенням функції одного або більше органів.
У формуванні клінічної гетерогенності ССД велике значення мають стать і вік хворих. При розвитку ССД у чоловіків переважає гострий чи підгострий перебіг, дифузна клінічна форма. Удвічі частіше, ніж у жінок (зокрема в дебюті), спостерігаються поширені шкірні, здебільшого індуративні, зміни із супутньою суглобово-м’язовою симптоматикою (частий розвиток контрактур та асоціація із синдромом поліміозиту). СР у дебюті ССД у чоловіків реєструють лише в 35% випадків, проте загалом спостерігаються типові виражені порушення мікроциркуляції. Характерними вісцеральними проявами є ураження серця з порушеннями ритму та провідності, інтерстиційний легеневий фіброз та легенева гіпертензія.
При ССД із пізнім дебютом (в осіб віком понад 50 років) переважно спостерігаються лімітовані шкірні прояви (75%) – частіше не індуративні, а атрофічні, при цьому характерними є тяжкі ішемічні ураження кінцівок, частий та швидкий розвиток вісцеральної патології (прогресуюче ураження міокарда із систоло-діастолічною дисфункцією лівого шлуночка, залученням клапанів серця, легень – із швидким розвитком легеневої гіпертензії – в перші 3 роки хвороби). Більш рідкісним є поєднання ССД з іншими СЗСТ та синдромом Шегрена, більш частим – з ОА. Ґенез судинно-вісцеральних уражень змішаний: у його основі лежать як склеродермічні мікроциркуляторні порушення та фіброз, так і супутні стани – артеріальна гіпертензія, атеросклероз. Таким чином, при пізньому дебюті ССД лімітований шкірний синдром втрачає своє класифікаційне та прогностичне значення, оскільки нерідко поєднується з гострим перебігом та ураженням життєво важливих органів.
Список літератури знаходиться в редакції.