


23 серпня, 2015
Мозаичный вариант частичной трисомии по длинному плечу хромосомы 22 у ребенка с врожденным пороком сердца

Известно, что примерно 0,5% всех новорожденных имеют хромосомные аномалии. Хромосомные перестройки являются одной из многочисленных причин нарушений нормального пре- и постнатального развития. Если полные трисомии и моносомии большинства хромосом приводят к гибели плода уже в пренатальный период, то частичные трисомии и моносомии постнатально встречаются достаточно часто. Отклонения от нормального развития в этих случаях определяются, главным образом, хромосомными районами, которые представлены аномальным числом.
В литературе описаны случаи частичной тетрасомии/трисомии длиного плеча хромосомы 22, являющейся редкой хромосомопатией, обусловливающей развитие синдрома «кошачьего глаза» (англ. Сat eye syndrome, СЕS) (код в ОМIМ1 115470) [26, 32, 44, 49]. Частота встречаемости данного заболевания среди живoрожденных варьирует от 1:50 000 до 1:150 000 [14, 55], а в случае спонтанных абортов составляет 10%. Дисбаланс хромосомного материала при частичной тетрасомии/трисомии хромосомы 22 приводит к нарушению онтогенеза, опосредуя в большинстве случаев гибель плода во внутриутробном периоде; летальность на первом году жизни связана преимущественно с кардиоваскулярными мальформациями либо с интеркуррентными инфекциями. Тем не менее, минорные фенотипические варианты могут существенно не влиять на продолжительность жизни.
Согласно данным литературы, при СЕS отмечают фенотипическую и цитогенетическую вариабельность [3, 6, 10, 37, 46]. Клиническое разнообразие проявлений синдрома «кошачьего глаза» затрудняет диагностику этого заболевания при медико-генетическом консультировании и требует обязательного цитогенетического, а в случае необходимости и молекулярно-цитогенетического исследования. Описание клинической картины новых случаев СЕS необходимо для изучения фено-кариотипической корреляции и определения диагностической значимости сопутствующих аномалий при данной хромосомопатии.
Цель работы – уточнение диагноза у ребенка с кардиоваскулярной мальформацией и признаками дисэмбриогенеза.
Материалы и методы исследования
Цитогенетическую диагностику осуществляли на препаратах метафазных хромосом, которые готовили из 72-часовой культуры лимфоцитов периферической крови, стимулированных фитогемагглютинином, по общепринятому методу [4]. Хромосомы идентифицировали после дифференциального окрашивания (GTG- и С-методами) при увеличении ×1000. Идентификацию хромосом выполняли в соответствии с описанием стандартного кариотипа человека.
Результаты исследования и их обсуждение
Пробанд К. – годовалая девочка, родилась от 4-й беременности; 1-я закончилась рождением здорового мальчика на 40-й неделе; 2-я – самопроизвольным выкидышем на 9-10-й неделе; 3-я – рождением здорового мальчика на 36-37-й неделе. Родители соматически здоровы, брак неродственный, родословная не отягощена. 4-я беременность протекала с явлениями гипертрофии плаценты и гипотрофии плода, маловодия (по данным ультразвукового исследования [УЗИ] во втором и третьем триместрах соответственно). Роды самостоятельные, однократное обвитие пуповиной вокруг шеи, асфиксия. Девочка родилась на 42-ой неделе с массой тела 3060 г, длиной 55 см. После рождения диагностирован врожденный порок сердца (ВПС): тотальный аномальный дренаж легочных вен (инфакардиальная форма); вторичный дефект межпредсердной перегородки; добавочная верхняя полая вена. Ребенок был прооперирован в Научно-практическом центре детской кардиологии и кардиохирургии Министерства здравоохранения Украины г. Киева. Кардиологический статус соответствует состоянию после оперативной коррекции ВПС.
При клиническом осмотре пробанда на момент обследования: брахицефальная форма головы с выступающим лбом, глубоко посаженными глазами; широкая, вдавленная переносица; деформированные ушные раковины; преаурикулярные фистулы; клинодактилия мизинцев; кривошея; низкопосаженные первые пальцы кистей. Ходит с опорой, активно ползает, произносит слоги. УЗИ органов брюшной полости патологии не выявило. Особенности фенотипа ребенка позволили предположить, что причиной нарушений онтогенеза является хромосомная аномалия. Поэтому для верификации диагноза провели кариотипирование. При цитогенетическом исследовании у ребенка обнаружен мозаичный кариотип с наличием клона клеток с частичной трисомией по длинному плечу хромосомы 22 – mos 47,XX,+del(22)(q11.2)[32]/46,XX[22] (рис. 1).
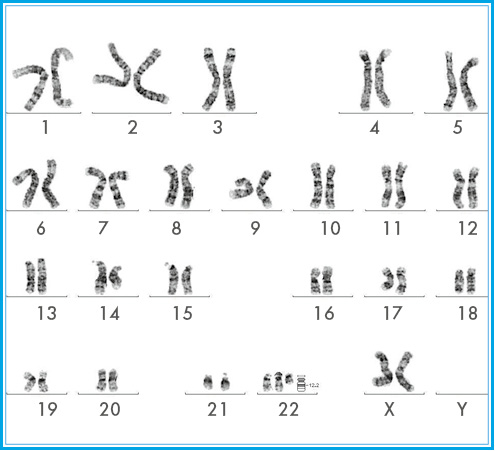 Рисунок 1. Метафазная пластинка с набором хромосом 47,XX,+del(22)(q11.2) (GTG-метод окрашивания, ок. 10 × об. 100)
Рисунок 1. Метафазная пластинка с набором хромосом 47,XX,+del(22)(q11.2) (GTG-метод окрашивания, ок. 10 × об. 100)Кариотипы родителей и двух сибсов в норме: 46,ХХ у матери; 46,ХУ у отца и сибсов; таким образом, хромосомная патология у пробанда возникла de novo. Синдром частичной тетрасомии/трисомии длиного плеча хромосомы 22 был подробно описан в 70-х годах прошлого века и из-за вертикальной локализации колобомы радужной оболочки назван синдромом «кошачьего глаза».
Современные представления о синдроме кошачьего глаза постулируют уникальность хромосомной аберрации при CES, заключающуюся в высокой вариабельности клинической экспрессии патологии [49, 35]. В сочетании с редкостью синдрома это предполагает незавершенность систематизации фенотипических презентаций, отличающихся крайним полиморфизмом даже в пределах одной семьи [8, 45].
Классическая триада предусматривает наличие:
- мальформаций ушных раковин в виде преаурикулярных сосочков и/либо фистул;
- анальной атрезии;
- колобомы радужной оболочки, преимущественно вертикальной.
Однако приблизительно для 60% пациентов последние структурные компоненты нехарактерны и замещены иными различными вариациями [5, 42]. Прослеживается полиорганность и полисистемность поражений с формированием индивидуальных анатомо-морфологических кластеров, от минорных до обширных и тяжелых. Высокое прогностическое значение имеют ассоциированные с CES врожденные пороки развития кардиоваскулярной системы при доминировании в структуре тотального аномального дренажа легочных вен (англ. Total Anomalous Pulmonary Venous Return, TAPVR) [8, 25] и аномалий развития дуги аорты (перерывы типа В) [54]. Несколько реже упоминаются септальные дефекты (вторичные дефекты межпредсердной перегородки и перимембранозные дефекты межжелудочковой перегородки, гипоплазия правого желудочка, атрезия легочной артерии, открытый артериальный проток) [51]. В дальнейшем изучении нуждаются единичные литературные сведения об обнаружении у пациентов репродуктивного возраста систолической дисфункции левого желудочка и умеренной клапанной регургитации [8]. Практически тотально распространен краниофациальный дисморфизм (вышеупомянутые особенности анатомии ушных раковин, неправильная форма черепа, низкая линия роста волос на голове, широкая переносица, глазной гипертелоризм, тонкая верхняя губа, ретрогнатия, мандибулярная гипоплазия, расщелины неба, опущение век, эпикант, страбизм в рамках офтальмологического синдрома Дуэйна, асимметрия лица и т. д.) [8, 42]. Аурикулярные дефекты могут сочетаться с атрезией наружного слухового прохода, гипоплазией барабанной полости, приводящими к сурдологическим расстройствам кондуктивного типа [27]. Особого внимания заслуживает неврологический паттерн индивидов с CES. Тяжелая задержка психомоторного развития не характерна (мировая литература содержит описание единичных случаев), в то время как легкие либо умеренные формы когнитивной и моторной ретардации в той или иной мере присущи приблизительно каждому третьему пациенту [27]. Нередко присутствуют пупочные грыжи, урогенитальные аномалии (гипоплазия почек, крипторхизм, др.) [13, 29, 31]. Незначительно представлены аспления либо добавочная селезенка, гиперинсулинемия, иммунодефицитные состояния, тератомы [12, 25, 53].
В настоящее время рассматривается зависимость фенотипической презентации от определенных генов региона хромосомы 22, связанного с развитием CES. Предполагается, что ген CECR1 способен опосредовать возникновение кардиальных и лицевых пороков, ген CECR2 может быть вовлечен в хроматидное ремоделирование, а гены MIL1 и BID, вероятно, связаны с нарушениями физического и ментального развития [25].
Высказывается предположение, что при мозаичных формах заболевания наличие нормального клеточного клона коррелирует с меньшей степенью выраженности клинической экспрессии, однако подобное утверждение нуждается в дальнейших исследованиях.
G. Schachenmann и соавторы [48] первыми указали на ассоциацию между наличием у пациентов колобомы радужной оболочки и атрезии ануса и присутствием в их кариотипе дополнительной маркерной хромосомы. Впоследствии было установлено, что дополнительная хромосома является производной хромосомы 22. Согласно данным литературы, синдром СЕS характеризуется не только вариабельностью фенотипических проявлений, но и разнообразием цитогенетических характеристик. Так, клинические проявления синдрома СЕS наблюдают при:
- частичной тетрасомии, обусловленной наличием в кариотипе дополнительной бисателлитной (+idic22 (pter→ q11.2::q11.2 → pter) [6, 14, 15, 31] или кольцевой (іdісr(22)(p11.2q11.21) [20, 22] маркерной хромосомы;
- частичной трисомии, являющейся результатом наличия дополнительной делетированной хромосомы 22 (del(22)(q11.2)) [3, 9, 28] или дупликации (прямой или инвертированной) [30, 34, 37, 43].
Описаны мозаичные [22, 25, 33] и транслокационные [2, 11, 23] варианты кариотипа при СЕS.
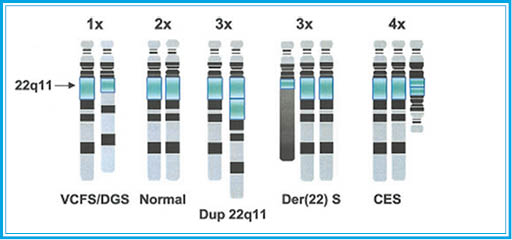 Рисунок 2. Схематическое изображение перестроек, затрагивающих район 22q11. Над парами гомологичных хромосом 22 указано число копий q11, под – синдромы, обусловленные конкретной реорганизацией 22q11.2 (E. Heather et al., 2002)
Рисунок 2. Схематическое изображение перестроек, затрагивающих район 22q11. Над парами гомологичных хромосом 22 указано число копий q11, под – синдромы, обусловленные конкретной реорганизацией 22q11.2 (E. Heather et al., 2002)Следует заметить, что в районе 22q11 локализовано несколько «горячих точек», характеризующихся район-специфичными низкокопийными повторами (англ. Low Copy Repeats, LCRs), рекомбинации между которыми обусловливают реорганизацию хромосомы [7, 17, 18, 26, 38, 39, 41, 47]. Известно, что наряду с хромосомными перестройками, ведущими к частичной тетрасомии/трисомии 22q11.2, ассоциированной с CES, в районе 22q11.2 возможны микроделеции, обусловливающие синдромы САТСН 22 (аббревиатура латинских названий основных симптомов заболевания: Cardiac defects [пороки сердца], Abnormal facies [лицевые аномалии], Thymus hypoplasia [гипоплазия вилочковой железы], Cleft palate [небно-глоточная недостаточность], Hypocalcemia [снижение уровня кальция в сыворотке], del 22 [делеция района q11.2 хромосомы 22]), в том числе синдром Ди-Джорджи [1, 16, 52]. Необходимо отметить, что район 22q11.2 – это также место перестройки и при транслокации t(11;22). В семьях, где один из супругов является носителем такой транслокации, возможны потомки с аномальным кариотипом 47,+der(22)t(11;22)(q24.3;q11.2), т. е. с трисомией по участкам 22pter→22q11.2 и 11q24.3 → qter (der(22) синдром / синдром Эммануэль, код в ОМІМ 609029) и клинической презентацией, во многом сходной с таковой при синдроме «кошачьего глаза» [19, 21, 40, 50, 56] (pис. 2).
Представленный нами случай согласуется с данными литературы [10] о вариабельности клинических признаков синдрома «кошачьего глаза». Не исключено, что наличие в кариотипе клона клеток с нормальным набором хромосом внесло свой вклад в отсутствие у пробанда таких характерных для СES фенотипических признаков, как колобома радужной оболочки и аноректальные аномалии. В то же время вариант ВПС и ряд мальформаций, отмеченные у пробанда, являются характерными для данного заболевания.
Всестороннее изучение этого случая и планируемое длительное наблюдение за развитием пробанда позволит определить влияние дополнительного хромосомного материала на соматические, психические и социальные особенности ребенка.
Итак, в результате обследования у пробанда установлена редкая хромосомопатия – мозаичная форма частичной трисомии по участку 22q11.2. Проведенные исследования позволили своевременно поставить точный диагноз, уточнить клинический прогноз с предикативными аспектами возможных нарушений развития индивидуума и разработать тактику коррекции этих нарушений.
В заключение нужно отметить, что дальнейшее накопление и систематизация данных о гено-фенотипических корреляциях при СES будет способствовать улучшению дифференциальной диагностики заболевания и, следовательно, повышению качества медико-генетического обследования.
Литература
1. Антоненко В.Г. Синдромы САТСН22. Del22: вариабельность клинических проявлений / В.Г. Антоненко, Л.Я. Левина, Л.М. Константинова // Российский вестник перинаталогии и педиатрии.- 2000 - №3 - C.47-52.
2. Барышев Ю.И. Транслокационный вариант трисомии 22 у ребенка с умственной отсталостью / Ю.И. Барышев, П.В. Новиков, С.Г. Ворсанова и др. // Вопросы охраны материнства и детства – 1989. – T. 34, № 2. – C. 71-74.
3. Горовенко Н.Г. Випадок часткової трисомії хромосоми 22 у дитини без колобоми райдужки / Н.Г. Горовенко, Н.О. Тищенко, Т.Е. Зерова-Любимова, О.Г. Євсеєнкова // Збірник наукових праць співробітників КМАПО ім. П.Л. Шупика. – Вип. 13, книга 5. – Київ, 2004. – C. 74-78.
4. Зерова-Любимова Т.Е. Цитогенетичні методи дослідження хромосом людини (методичні рекомендації) / Т.Е. Зерова-Любимова, Н.Г. Горовенко – К., – 2003. – 23 с.
5. Allotey J. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy / J. Allotey, F. Lacaille, M.M. Lees et al // J.Pediatr. Surg. – 2008 – V. 43 (9) – P. 1736-1740.
6. Avior G. Associated brachial cleft anomalies in the cat eye syndrome / G. Avior, A. Derowe, D.M. Fliss et al. // Harefuah. – 2007 – V. 146 (2). – P. 99-101; P. 166-167.
7. Babcock M. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution / M. Babcock, A. Pavlicek, El. Spiteri, C.D. Kashork et al // Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1549503.
8. Belangelo S.I. Wide clinical variability in cat eye syndrome patients: four non-related patients and three patients from the same family / S.I. Belangero, A.N. Pacanaro, F.T / Bellucco et al // Cytoqenet. Genome. Res. – 2012. – V. 138 (1). – P. 5-10/
9. Belien V. Partial trisomy of chromosome 22 resulting from a supernumerary marker chromosome 22 in a child with features of cat eye syndrome / V. Belien, M. Gerard-Blanluet, S. Serero et al // Am. J. Med. Genet. – 2008 – V. 146A (14) – P. 1871-1874.
10. Berends M.J. Phenotypic variability of cat-eye syndrome / M.J. Berends, G. Tan-Sindhunata, B. Leegte, A.J. van Essen // Genet. Couns. – 2001 – V. 12 (1) – P. 23-34.
11. Buhler E.M. Cat-eye syndrome, a partial trisomy 22 // E.M. Buhler, K. Mehes, H. Muller, G.R. Stalder / Humangenetik. – 1972 – V. 15 – P. 150-162.
12. Chellapandian D. Anatomical asplenia in cat eye syndrome: an expansion of the disease spectrum / D. Chellapandian, A. Schneider // Pediatrics. – 2013 – Article ID 218124, 4 pages http://dx.doi.org/10.1155/2013/218124
13. Chen C.P. Prenatal diagnosis and molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 22 associated with cat eye syndrome / C.P. Chen, T.M. Ko, Y.Y. Chen et al // Gene. – 2013 – V. 527 (1) – P. 384-388.
14. Cullen P. Association of familial Duane anomaly and urogenital abnormalities with a bisatellited marker derived from chromosome 22 / P. Cullen, C.S. Rodgers, D.F. Callen et al // Am. J. Med. Genet. – 1993. – V. 47 – P. 925-930.
15. Córdova-Fletes C. A de novo sSMC(22) characterized by high-resolution arrays in a girl with cat-eye Syndrome without coloboma / C. Córdova-Fletes, M.G. Domínguez, A. Vázquez-Cárdenas, L.E. Figuera, V.A. Neira, A. Rojas-Martínez, R. Ortiz-López // Mol. Syndromol. – 2012. – V. 3 – P. 131-135.
16. DiGeorge A. A new concept of the cellular basis of immunity / A. DiGeorge // J. Pediatr. – 1965 – V. 67 – P. 907.
17. Edelman L. Low-copy repeats mediate the common 3-Mb deletion in patients with Velo-cardio-facial syndrome / L. Edelman, R.K. Pandit., B.E. Morrow // Am. J. Hum. Genet. – 1999 – V. 64 – P. 1076-1086.
18. Edelman L. A common molecular basis for rearrangement disordeson chromosome 22q11 / L. Edelman, R.K. Pandita, E. Spiteri et al // Hum. Mol. Genet. – 1999 – V. 8 – P. 1157-1167.
19. Emanuel B.S. The 22q11.2 deletion syndrome / B.S. Emanuel, D. McDonald-McGinn, S. C. Saitta, E. H. Zackai // Adv. Pediatr. – 2001. – V. 48 – P. 39-73.
20. El-Shanti H. A three generation minute supernumerary ring 22: association with cat-eye syndrome / H. El-Shanti, H. Hulseberg, J.C. Murray, S.R. Patil // Am J. Hum. Genet. Suppl – 1993. – 53: A126.
21. Fraccaro M. The 11q;22q translocation: a European collaborative analysis of 43 cases / M. Fraccaro, J. Lindsten, C.E. Ford, L. Iselius // Hum. Genet. – 1980 – V. 56. – P. 21-51.
22. Frizzley J.K. Ring 22 duplication/deletion mosaicism: clinical, cytogenetic, and molecular characterisation / J.K. Frizzley, M.J. Stephan, A. Lamb et al // J. Med. Genet. – 1999 – V. 36 (3) – P. 237-241.
23. Gabarrón J., Pseudoisodicentric bisatellited extra marker chromosome (tetrasomy 22pter----q11, trisomy Yqh), derived from a maternal Y/22 translocation. Association between this tetrasomy and "cat eye" phenotypical features / J. Gabarrón, G. Glover, A. Jimene, E. Lamata // Clin. Genet. – 1985. – V. 28 (6). – P. 509-515.
24. Gadji M. Prenatal diaqnosis and molecular characterizationof two constituonal rinqs derived from one chromosome 22 / M. Gadji, R. Krabchi, A. Aboura et al // Am J Med. Genet. – 2011. – V. 155A – P. 430-433.
25. Haltrich I. A de novo atypical ring sSMC(22) characterizet by arry CGH in a boy with cat-eye syndrome / I. Haltrich, H.Pico, E. Kiss et al // Molecular cytogenetic. – 2014. – V. 7. – P. 37. http:www.molecularcytoqenetics.orq/content/7/1/37
26. Jalali G.R. Detailed Analysis of 22q11.2 With a High Density MLPA Probe Set / G.R. Jalali, J.A.S. Vorstman, A. Errami et al // Hum Mutat. – 2008. – V. 29 (3). – P. 433-440.
27. Jedraszak G. Severe psychomotor delay in a severe presentation of cat-eye syndrome / Jedraszak G, Receveur A, Andrieux J, Mathieu-Dramard M, Copin H, Morin G // Case Rep. Genet. – 2015. – V. 2015. – P. 943905. doi: 10.1155/2015/943905.
28. Jezela-Stanek A. Trisomy 22pter-q12.3 presenting with hepatic dysfunction variability of cat-eye syndrome / A. Jezela-Stanek, A. Dobrzarinska, M.D. Gasiorek et al // Clin. Dysmorphol. – 2009 – V. 8 (1). – P. 13-17.
29. Knijnenbura J. A 600 kb triplication in the cat eye syndrome critical region causes anorectal, renal and preauricular anomalies in a three-generation family / J. Knijnenbura, Y. van Bever , L.O. Hulsman et al // Eur. J. Hum. Genet. – 2012. – V. 20 (9) – P. 986-969.
30. Knoll J.H.M. Interstitial duplication of proximal 22q: phenotypic overlap with cat eye syndrome / J.H.M. Knoll, A. Asamoah, B.A. Pletcher, J. Wagstaff // Am. J. Med. Genet. – 1995 – V. 55. – P. 221-224.
31. Ko J.M. Partial tetrasomy of chromosome 22q11.1 resulting from a supernumerary isodicentric marker chromosome in a boy with cat-eye syndrome / M.J. Ko, J.M. Kim, K.S. Pai et al // J. Korean Med. Sci. – 2010 – V. 25 (12) – P. 1798-801.
32. Kollárová A. The Schmid – Fraccaro syndrome / A. Kollárová, N. Misovicová, V. Mális // Cesk. Slov. Oftalmol. – 1999. – V. 55 (6). – P. 362-366.
33. Kvarnung M. Inherited mosaicism for the supernumerary marker chromosome in cat eye syndrome: inter- and intra-individual variation and correlation to the phenotype / M. Kvarnung, A. Lindstand, H. Malmgren et al //Am J. Med. Genet. – 2012. – V. 158A (5). – P. 1111-1117.
34. Lindsay E.A. De novo tandem duplication of chromosome segment 22q11-q12: clinical, cytogenetic, and molecular characterization / E.A. Lindsay, L.G. Shaffer, R. Carrozzo et al // Am. J. Med. Genet. – 1995. – V. 56. – P. 296-299.
35. Matsumoto R. Cat-eye syndrome with isolated idiopathic hypogonadotropic hypogonadism / R. Matsumoto, C. Shimizu, S. Nagai et al // Intern. Med. – 2005. – V. 44 (10). – P. 1069-1073.
36. Mears A.J. Minute Supernumerary ring chromosome 22 associated with cat eye syndrome: Further delineation of the critical region / A.J. Mears, H. El-Shanti, J.C. Murray et al // Am. J. Hum. Genet. – 1995. – V. 57. – P. 667-673.
37. Meins A.J. Partial trisomy of chromosome 22 resulting from an interstitial duplication of 22q11.2 in a child with typicalcat eye syndrome / M. Meins, P. Burfeind, S. Motsch, et al / J. Med. – 2003. – V. 40. – P. e62.
38. McDermid H.E. Long-range mapping and construction of a YAC contig within the Cat eye syndrome critical region / H.E. Mc Dermid, K.E. Mc Taggart, M. Ali Riazi et al. // Genome Res. – 1996. – V. 6. – P. 1149-11599.
39. McTaggart K.E. Cat Eye Syndrome chromosome breakpoint clustering: identification of tho intervals also associated with 22q11 deletion syndrome / K.E. McTaggart, M.L. Budarf, D.A. Drisoll et al // Cytogenet. Cell. Genet. – 1998. – V. 81. – P. 222-228.
40. Prieto J.C. Phenotypic expansion of the supernumerary derivative (22) chromosome syndrome: VACTERL and Hirschsprung's disease / J.C. Prieto, N.M. Garcia, F.F. Elder et al // J. Pediatr. Surg. – 2007. – V. 42 (11). – P. 1928-1932.
41. Puech A. Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization / A. Puech, B. Saint-Jore, B. Funke et al // Proc. Natl. Acad. Sci. USA. – 1997. – V. 94 – P. 14608-14613.
42. Quintero-Rivera F. Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry / F. Quintero-Rivera, J.A. Martinez-Agosto // Am. J. Med. Genet. A. – 2013. – V. 161A (8). – P. 1985-1991.
43. Reiss J.A. Tandem duplication of proximal 22q: a cause of cat-eye syndrome / J.A. Reiss, R.G. Weleber, M.G. Brown RE. et al // Am. J. Med. Genet. – 1985. – V. 20. – P. 165-171.
44. Romagna E.S. Schmid-Fraccaro syndrome: severe neurologic features / E.S. Romagna, M.S. Appel da Silva, P.A. Ballardin // Pediatr. Neurol. – 2010. – V. 42 (2). – P.151-153.
45. Rosa R.F. Clinical characteristics of a sample of patients with cat eye syndrome / R.F. Rosa, R. Mombach, P. R. Zen et al / Rev. Assoc. Med. Bras. – 2010. – V. 56 (4). – P. 462-5.46.
46. Rosias P.R. Phenotypic variability of the cat eye syndrome. Case report and review of the literature / P.R. Rosias, J.M. Sijstermans, P.M. Theunissen et al. // Genet. Couns. – 2001. – V. 12. – P. 273-282.
47. Shaikh T.N. Chromosome 22-specific low copy Repeats and the 22q11.2 deletion syndrome: Genomic organisation and deletion endpoint analysis / T.N. Shaikh, H. Kurahashi, S.C. Saitta et al // Hum. Mol. Genet. – 2000. – V. 9 – P. 489-501.
48. Schachenmann G. Chromosomes in coloboma and anal atresia / G. Schachenmann, W. Schmid, M. Fraccaro et al // The Lancet. – 1965 – V. 286, № 7406. – P. 290-299.
49. Schinzel A. Catalogue of unbalanse chromosome aberrations in man / A. Schinzel // Berlin, New York, de Gruyter – 2001. – 966 p.
50. Schinzel A. Incomplete trisomy 22. I. Familial 11/22 translocation with 3:1 meiotic disjunction: delineation of a common clinical picture and report of nine new cases from six familie / A. Schinzel , W. Schmid, P. Maur et al / Hum. Genet. – 1981 – V. 56. – P. 249-262.
51. Sharma D. Cat eye syndrome / D. Sharma, S. Murki, T. Pratar, M. Vasikarla // B. M. J. Case Rep. – 2014. – May 19. pii: bcr2014203923. doi: 10.1136/bcr-2014-203923.
52. Shprintzen R.J. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome / R.J. Shprintzen, R.B. Goldberg, M.L. Lewin // Cleft. Palate J. – 1978. – V. 15. – P. 56-62.
53. Tzetis M. An unusual case of Cat-Eye syndrome phenotype and extragonadal mature teratoma: review of the literature / M. Tzetis, K. Stefanaki, A. Surmou et al // Clin. Mol. Teratol. – 2012. – V. 94 (7). – P. 561-566.
54. Tsai M.C. / Type B Interrupted Aortic Arch and Hydrocephalus Associated with Mosaicism of a 1.37 Mb Amplified Cat Eye Syndrome Critical Region // Tsai M.C., Chou Y.Y., Wang J.N., Wu J.M., Huang C.C., Kuo P.L., Tsai Y.S. Pediatr. Neonatol. – 2015. – Jan. 3. pii: S1875-9572(14)00204-6. doi: 10.1016/j.pedneo.2014.10.009
55. Win T.N. Duane syndrome associated with the Cat Eye syndrome: a case report / T.N. Win, S. Roberts, D. Laws // Eye – 2007. – V. 21, № 2. – P. 289-291.
56. Zackai E.H. Site-specific reciprocal translocation, t(11;22)(q23;q11), in several unrelated families with 3:1 meiotic disjunction / E.H. Zackai, B.S. Emanuel // Am. J. Med. Genet. – 1980. – V. 7. – P. 507-521.
1 OMIM™ (англ. Online Mendelian Inheritance in Man – Менделевская наследственность у человека онлайн) – проектная база данных Университета Джона Хопкинса (англ. Johns Hopkins University) в США, представляющая собой каталог заболеваний, ассоциированных с генетическим компонентом.