


1 квітня, 2015
Биосимиляры: эффективность и безопасность, требующие доказательств
По материалам научно-практической конференции
 В рамках научно-практической конференции «Инновационные технологии лечения в ревматологии с позиций доказательной медицины», проходившей 30-31 октября 2014 года в г. Киеве, состоялось заседание экспертного совета «Биосимиляры – объективизация биологической терапии ревматических заболеваний». Основные положения некоторых из прозвучавших докладов представлены ниже.
В рамках научно-практической конференции «Инновационные технологии лечения в ревматологии с позиций доказательной медицины», проходившей 30-31 октября 2014 года в г. Киеве, состоялось заседание экспертного совета «Биосимиляры – объективизация биологической терапии ревматических заболеваний». Основные положения некоторых из прозвучавших докладов представлены ниже.
Академик НАМН Украины, президент Ассоциации ревматологов Украины, директор ГУ «ННЦ «Институт кардиологии им. Н.Д. Стражеско» НАМН Украины», доктор медицинских наук, профессор Владимир Николаевич Коваленко, открывая заседание, коснулся вопроса о возможности и правомерности постановки знака равенства между оригинальными биопрепаратами и биосимилярами. Ответ на этот вопрос был получен в ходе обсуждения различий в процессах производства этих молекул, а также аспектов эффективности и безопасности применения биосимиляров.
Начальник Департамента пострегистрационного надзора ГП «Государственный экспертный центр МЗ Украины» Елена Валерьевна Матвеева дала определение биосимилярам, остановившись на аспектах безопасности их применения. Биосимиляр – копия уже зарегистрированного (референтного) биопрепарата с доказанным подобием на основе всестороннего сопоставления физико-химических характеристик, эффективности и безопасности. В англоязычной литературе встречаются разные названия биосимиляров: similar biological medicinal product (EMA), follow on protein product (FDA), similar biotherapeutic product (ВОЗ).
Биологический и химический препараты отличаются по ряду показателей (табл.).
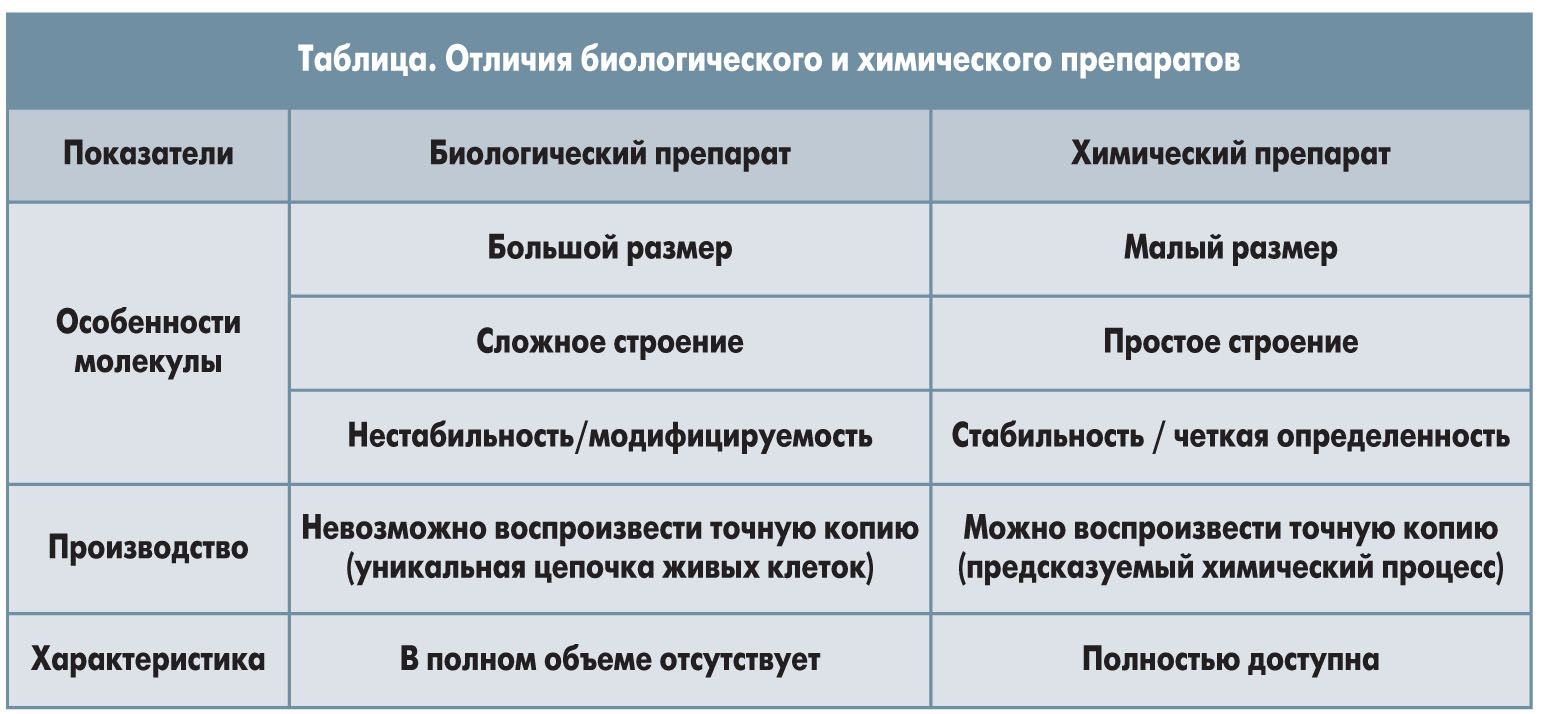
Ввиду неизбежных отличий в производственных процессах (применение разных систем экспрессии целевого гена, методов ферментации и очистки, вспомогательных веществ) профиль качества биосимиляров и референтных биопрепаратов не может быть строго идентичным. Этот факт принят научным сообществом и регуляторными органами как данность, начиная с момента внедрения технологий для производства биопрепаратов. Таким образом, биосимиляры схожи с оригинальным препаратом, но не идентичны ему, и в этом заключается их отличие от генериков.
В Евросоюзе любой биосимиляр, представленный на регистрацию, оценивают с помощью всестороннего сравнения с референтным средством, включая сопоставление профиля качества и данных неклинических/клинических исследований, цель которых заключается в подтверждении сходности физико-химических характеристик, безопасности и эффективности. Столь же всесторонне оценивают и оригинальный биопрепарат, если в процесс его производства были внесены какие-либо изменения. На протяжении жизненного цикла лекарственного средства изменения вносятся достаточно часто, например, чтобы улучшить его качество или увеличить объемы производства. Это, в свою очередь, обусловливает необходимость мониторинга профиля безопасности биопрепарата в течение его жизненного цикла.
К серьезным последствиям может привести внесение даже, на первый взгляд, незначительных изменений в технологии производства препарата. Подтверждением этого служит пример с препаратом Эпрекс (эпоэтин альфа).
Одним из показаний к его применению является лечение вторичной анемии у пациентов с хронической почечной недостаточностью. Внесение небольшой корректировки в технологию производства Эпрекса привело к индукции выработки у некоторых больных нейтрализующих антител, причем не только к препарату, но и к эндогенному эритропоэтину. Результатом этого стало развитие аплазии костного мозга с поражением клеток-предшественников эритропоэза. Единой точки зрения, которая бы объяснила возникшую клиническую ситуацию, не существует. Согласно так называемой рабочей гипотезе, конформация молекулы препарата произошла вследствие взаимодействия между непокрытыми резиновыми пробками и новым стабилизатором.
Основной проблемой при использовании биопрепаратов, в том числе биосимиляров, является их иммуногенность. Иммуногенный потенциал биопрепарата невозможно предсказать с помощью химического или структурного анализа. Тем не менее известно несколько факторов, повышающих иммуногенность: наличие примесей в конечном продукте, структурные изменения в результате производственного процесса и/или условий хранения, иммунный статус пациента, генетический фон, характер заболевания. Проблема идентификации иммуногенности состоит в том, что она далеко не всегда проявляется в виде побочных реакций. Главным ее последствием является недостаточная эффективность биопрепарата, которую трудно оценить.
Учитывая это, с точки зрения фармаконадзора нецелесообразной является замена биопрепарата на биосимиляр без достаточных на то оснований, поскольку это может привести к развитию иммуногенности. Решение о замене биопрепарата должно приниматься только высококвалифицированными специалистами. Производители биосимиляров и врачи обязаны предоставлять пациентам и фармацевтам всю известную им информацию о рисках, связанных с переходом с оригинального биопрепарата на биосимиляр.
При исходном определении взаимозаменяемости и принятии решения о замене учитываются следующие факторы:
• относящиеся к лекарственному средству – заключение о сопоставимости препарата, сравнительные данные доклинических и клинических исследований, опыт применения биосимиляров и оригинального биопрепарата, профиль безопасности последнего, спецификация планов управления рисками;
• относящиеся к пациенту – терапевтические показания, опыт лечения данным препаратом в прошлом, мониторинг состояния больного, информация о нем.
Ключевым вопросом при осуществлении фармаконадзора за биосимилярами является четкая идентификация лекарственного средства по торговому названию, внешнему виду первичной и вторичной упаковки, которые должны отличаться от таковых референтного биопрепарата. Кроме того, важно знать номер серии, дату выпуска, показания к применению биосимиляра.
Обязательным условием присутствия биосимиляра на рынке является осуществление фармаконадзора. При этом, как правило, используется потенциал рутинного фармаконадзора, что подразумевает сбор сообщений о случаях развития побочных реакций с помощью метода спонтанного рапортирования, предоставление периодически обновляемых отчетов по безопасности (Periodic Safety Update Report – PSUR), работу с сигналом и др.
Однако метод спонтанного рапортирования имеет ряд недостатков, включая отсутствие данных об экспозиции пациентов, трудности при определении причинно-следственной связи. Например, было установлено, что у лиц, страдающих воспалительными заболеваниями кишечника и принимающих ингибиторы фактора некроза опухоли альфа, повышается риск развития лимфомы. Однако эти пациенты зачастую одновременно принимают и другие иммуносупрессивные средства, поэтому относительно возникновения лимфомы не ясно, о побочном действии какого из препаратов следует сообщать. Еще больше осложняет оценку побочных реакций, особенно отсроченных, переход с оригинального биопрепарата на его аналог.
Несмотря на недостатки, метод спонтанных сообщений служит чрезвычайно важным инструментом для идентификации новых, редких и серьезных побочных реакций. Основная цель периодически обновляемого отчета по безопасности препарата – предоставить регуляторному органу имеющуюся информацию о безопасности и эффективности использования лекарственного средства во всех странах, где он разрешен для медицинского применения, а также оценку соотношения польза/риск. PSUR по биосимилярам имеет свои особенности. В нем внимание сосредоточено на безопасности других биопрепаратов с тем же активным веществом, а также детально проанализированы случаи иммуногенности (главным образом отсутствие эффекта). Для биосимиляра обязательно создается план управления рисками, что позволяет реализовать так называемый проактивный риск-менеджмент биосимиляров. План управления рисками (ПУР) является обязательной составляющей досье при регистрации биосимиляров в Евросоюзе. Этот документ четко структурирован. Ключевой частью ПУР является спецификация по безопасности, где должны быть представлены важные риски и отсутствующая информация. Под важными рисками подразумеваются такие, которые влияют на соотношение польза/риск, смещая его в сторону риска. В свою очередь, риски классифицируют на идентифицированные, когда существует четкая причинно-следственная связь между применением лекарственного средства и возникновением риска, и потенциальные, когда существует гипотеза, но весомые доказательства наличия взаимосвязи между применением лекарственного средства и возникновением риска отсутствуют. Спецификация по безопасности определяет активности по фармаконадзору, которые представляются в разделе ПУР «План по фармаконадзору», и по минимизации рисков, которые представляются в разделе ПУР «План минимизации рисков», если таковой приемлем. В плане по фармаконадзору заявитель описывает те методы, которые будут им использованы для дальнейшего выявления и оценки важных рисков и недостающей информации. Кроме обязательного рутинного фармаконадзора, заявитель биосимиляров должен обсудить и решить вопрос о необходимости дополнительных мероприятий по фармаконадзору, например пострегистрационных исследований. Такие мероприятия направлены на выявление и оценку иммуногенности (в том числе отсутствия эффективности), редких и серьезных побочных реакций, сбор информации о применении биосимиляра не по показаниям и др. Например, производителю вальтропина (референтный препарат – Хуматроп) необходимо исследовать реестр детей, отстающих в развитии от своего гестационного возраста. Это позволит получить данные о диабетогенном потенциале рекомбинантного человеческого гормона роста, риске возникновения гипотиреоза, формировании и клинической значимости антител, вырабатываемых к препарату.
В качестве еще одного примера может быть приведена ситуация с эпоэтином альфа (Hexal). В Евросоюзе данное лекарственное средство, в отличие от референтного препарата, не показано для лечения вторичной анемии при хронической почечной недостаточности. Поэтому компания-производитель обязалась провести маркетинговое исследование, цель которого заключается в мониторинге потенциального использования этого средства не по прямым показаниям.
Заместитель генерального директора ГП «Государственный экспертный центр МЗ Украины», доктор медицинских наук, профессор Татьяна Владимировна Талаева представила доклад «Биосимиляры. Что такое биосимиляры. Разница между генерическими препаратами и биосимилярами», в котором были обозначены основные подходы к регистрации генерических лекарственных средств и биосимиляров.
В последние десятилетия в фармацевтической отрасли отмечается стремительное развитие биотехнологий. С их помощью создаются принципиально новые лекарственные средства, которые более эффективно и избирательно действуют на патологические процессы. Однако это сопровождается возникновением ряда проблем, ключевой из которых является высокая стоимость таких препаратов, что обусловлено сложностью их разработки и производства. Стандартный путь решения проблемы относительно повышения доступности современных лекарственных препаратов для широких слоев населения предполагает замену оригинальных препаратов на их более дешевые воспроизведенные копии – генерики. Однако такой подход неприемлем, если речь идет о биосимилярах. Существенные отличия биотехнологических лекарственных средств от обычных синтетических препаратов, современные подходы, направленные на обеспечение их качества, эффективности и безопасности, требуют формирования нового мышления, подготовки новых высококвалифицированных кадров в области биотехнологий для изучения биоподобности, что предполагает полный отказ от «генерических» подходов.
Помимо биосимиляров также существуют неинновационные биопрепараты и биотехнологические средства II поколения. Неинновационный биопрепарат (me-too biologic/non-innovator biologic) разрабатывают самостоятельно и непосредственно (в поэтапных исследованиях) с референтным лекарственным средством не сравнивают. Такие продукты нельзя считать биосимилярами, поскольку для них не доказано подобие лицензированным средствам с точки зрения качества, безопасности и эффективности. Биотехнологические препараты II поколения, или биопревосходные средства (second generation biologic/biobetter), – продукты, которые были структурно и/или функционально изменены, для того чтобы оптимизировать клинический эффект или получить иное, отличающееся от оригинального, действие. Такие средства считаются отдельными (stand-alone products).
Примером служат препараты конъюгированных антител, которые используют одобренные моноклональные антитела либо новую лекарственную форму уже утвержденного средства. Биосимиляр и референтный препарат сходны молекулярно (имеют сходные массу, строение, аминокислотные состав и последовательность) и в производственном отношении (используется одинаковый метод получения активного вещества). В то же время при производстве биосимиляра и оригинального препарата применяют различные питательные среды, технологические циклы, способы очистки активной молекулы от компонентов цитоплазмы клетки-производителя. Количество и степень допустимых различий в каждом конкретном случае разные и зависят от многих факторов. Так, ПЕГ-интерферон альфа-2а содержит 11 лизиновых радикалов, любой из которых в силу особенностей технологического процесса может связаться с ПЕГ-про-теином. В результате образуются 11 позиционных изомеров. Однако лишь некоторые из них обладают биологической активностью и приемлемым уровнем иммуногенности.
Другой пример: ритуксимаб – генно-инженерные химерные моноклональные антитела, в которых соединены фрагменты иммуноглобулинов мыши и человека. Химерный ген, содержащий участки генов человека и мыши, внедряют в клетки яичника китайского хомячка. Культура этих клеток, помещенная в специальную питательную среду, вырабатывает химерный иммуноглобулин класса G1 (ритуксимаб). Молекула последнего состоит из 1328 аминокислот, поэтому теоретически возможно существование 285 млн структурных вариантов препарата.
Еще одним показательным примером служат низкомолекулярные гепарины (НМГ). Их производят методом химической или ферментативной деполимеризации «нефракционированного» гепарина, выделяемого из слизистой оболочки кишечника свиньи. НМГ различаются по химической структуре, методам получения и сродству к плазменным белкам. Параметры фармакокинетики для дальтепарина, надропарина, ревипарина и эноксапарина следующие: средняя молекулярная масса – 6000, 4300, 3900 и 4500 Да соответственно; период полувыведения (при внутривенном и подкожном введении) – 1,8-2,3 и 3,0-5,0 ч; 2,2-3,6 и 2,3-3,8 ч; 1,2-1,6 и 2,0-5,7 ч; 3,8-4,0 и 4,6-5,9 ч соответственно; биодоступность – 87, 98, 90, 91% соответственно. Имея сходный механизм действия, указанные НМГ обладают разной активностью (среднее отношение анти-Ха/анти-IIа активности – 2,7; 3,6; 3,5 и 3,8 соответственно). Следовательно, препараты НМГ не являются взаимозаменяемыми и имеют разные инструкции к применению (по утвержденным показаниям). Важно подчеркнуть, так как у производителя биосимиляра нет доступа к характеристикам оригинального биотехнологического продукта, он должен разработать собственные технологии получения рекомбинантной ДНК, штамм клеток, питательную среду для клеточной культуры, процесс очистки продукта, программу промежуточного контроля и пр.
Таким образом, различия между оригинальным препаратом и его биосимиляром могут быть очень велики, причем полное совпадение аминокислотного состава конечных продуктов не гарантирует их полной идентичности, степень которой априори предсказать нельзя.
Медицинские эксперты в области ревматологии и онкологии подчеркивают важность всестороннего исследования биосимиляров, включая дорегистрационную сравнительную оценку качества, безопасности и эффективности, а также строгий фармаконадзор в пострегистрационный период. Между тем современный контроль биотехнологических продуктов пока несовершенен. В частности, степень сходства биосимиляра с референтным препаратом не может выявляться обычными аналитическими методами. Например, структурные различия между оригинальным и биоподобным филграстимом не определяются хроматографически, но идентифицируются новой модификацией электрофореза. Наиболее часто для подтверждения строения и выяснения физико-химических свойств биопрепаратов используют такие методы, как: спектрофотометрия, анализ участков N- и С-концевой последовательности, пептидное картирование, электрофорез в полиакриламидном геле, капиллярный электрофорез, иммуноблоттинг, изоэлектрическое фокусирование, ядерно-магнитный резонанс, круговой дихроизм и пр. Сравнительную оценку оригинального биопрепарата и биосимиляра проводят на основе биоподобного подхода, подразумевающего пошаговый алгоритм демонстрации сопоставимости качества, безопасности и эффективности. При этом должное качество биосимиляра следует констатировать до начала сокращенной программы доклинических и клинических испытаний. Биоподобный подход применяют к высокоочищенным продуктам с хорошо известными характеристиками и одинаковыми с референтным препаратом дозой и путем введения. Биоподобный подход не распространяют на вакцины, аллергены, препараты генной и сомато-клеточной терапии, а также лекарственные средства, получаемые из плазмы. Исследования, доказывающие сопоставимость, необходимы в двух случаях. Во-первых, при значительных изменениях, вносимых производителем в собственный технологический процесс (препарат из новой партии сопоставляется с исходными спецификациями); во-вторых, если биопродукт заявлен как подобный оригинальному биопрепарату (биосимиляр сопоставляется с референтным препаратом). В последнем случае при идентификации значительных различий между препаратами заявленный продукт биосимиляром считать нельзя, однако его допустимо исследовать как отдельный препарат (без сравнения с референтным). Далее на основании полного досье средство регистрируют как неинновационный биотехнологический продукт.
В настоящее время увеличивается количество сообщений о различиях в качестве и отсутствии сходности между одинаковыми биопрепаратами разных производителей, с одной стороны, и оригинальным продуктом – с другой. Увеличилась частота развития истинной эритроцитарной аплазии у пациентов, получающих биосимиляры эритропоэтина. Так, в результате регуляторные органы Таиланда даже приостановили клиническое применение всех эритропоэтинов. Международная федерация фармацевтических производителей и ассоциаций (IFPMA) в своем программном заявлении (июль 2014 г.) предлагает ввести новый термин – «несопоставимые биотерапевтические продукты». Это понятие описывает препараты, которые предлагают как копии оригинальных, но, во-первых, не сравнивают с последними непосредственно по параметрам качества, безопасности и эффективности, а во-вторых, не регистрируют согласно процедуре, рекомендуемой для регистрации биосимиляров.
Для регуляторных органов IFPMA дает следующие рекомендации.
1. Устранить риск применения несопоставимых биотерапевтических продуктов путем пострегистрационной сравнительной оценки их качества, безопасности и эффективности по отношению к оригинальному препарату (в соответствии с рекомендациями ВОЗ).
2. Предусмотреть переходный период, в течение которого следует выполнить такую оценку для каждого несопоставимого продукта.
3. При идентификации значительных аналитических отличий, которые оказывают влияние на безопасность и эффективность средства, регуляторный орган должен дать возможность заявителю повторно зарегистрировать препарат как новый (на основании полного досье).
4. Чтобы гарантировать четкую идентификацию биопрепарата, который использовали для лечения, в процессе фармаконадзора обязательно использовать торговое название, наименование производителя и номер серии.
5. В инструкции по медицинскому применению несопоставимого биотерапевтического препарата следует предоставлять всю информацию, необходимую врачу для принятия решений.
Важнейшее условие сводится к тому, чтобы эта информация не дублировала инструкцию оригинального препарата, а основывалась на собственных клинических данных, подтверждающих показания к применению. Кроме того, целесообразно рассмотреть включение символа, который бы объяснял, что препарат не продемонстрировал сходство с оригинальным продуктом. Основными посылами IFPMA являются следующие: 1) любой зарегистрированный в стране биотехнологический препарат, предполагаемый как копия оригинального продукта, но не отвечающий критериям, которые установлены для биосимиляров (отсутствуют доказательства подобия качества, безопасности и эффективности), биосимиляром считаться не должен; 2) если заявитель не может предоставить все необходимые научные доказательства, позволяющие признать такой препарат биосимиляром, его оценку, служившую основанием для регистрации, регуляторные органы обязаны провести повторно; 3) в некоторых странах процесс повторной оценки может потребовать внесения изменений в регуляторные нормы (если они не соответствуют требованиям ВОЗ для биосимиляров и рДНК-продуктов).