


10 липня, 2016
Коллапсирующая гломерулопатия у ребенка с синдромом Галловея-Мовата

Клинический случай
Мальчик, 26 мес, поступил в отделение детской нефрологии Военно-медицинской академии с периорбитальным отеком и асцитом. Ранее наблюдался в другой клинике по поводу нистагма и микроцефалии.
Анамнез. Единственный ребенок в семье, родители здоровы. Семейный анамнез по заболеваниям почек не отягощен. Родился в срок с нормальными параметрами (вес 3250 г, рост 50 см, окружность головы 35 см), беременность протекала без осложнений.
Физикальный осмотр. Явная задержка психомоторного развития. Головку держит с 6 мес, не ходит и не говорит. На момент осмотра рост 85 см (25-й перцентиль), вес в отечном состоянии 12,7 кг (11 кг без отека, 10-25-й перцентиль), окружность головы 46 см (ниже 3-го перцентиля). Артериальная гипертензия (109/84 мм рт. ст.), выраженный горизонтальный и вертикальный нистагм. Лицевой дисмормизм отсутствует. Пальпация и перкуссия живота: выраженный асцит.
Лабораторные исследования: гемоглобин 18,5 г/дл, лейкоциты 9,6×109/л, тромбоциты 474×109/л; в сыворотке: мочевина 86 мг/дл, креатинин 0,7 мг/дл, низкие уровни альбумина (1,4 г/дл) и общего белка (3,6 г/дл), высокие уровни триглицеридов (1057 мг/дл) и общего холестерина (482 мг/дл); в анализе суточной мочи – протеинурия 55 мг/м2/ч, микроскопическая гематурия (3+) и альбуминурия (4+), органические кислоты в норме. Вирусные маркеры (HBsAg, анти-HCV, HIV, EBV, CMV и парвовирус) отрицательные. Комплемент сыворотки и антинуклеарные антитела нормальные.
Ультразвуковое исследование: почки нормальных размеров с повышенной кортикальной эхотекстурой и утратой кортикомедуллярной дифференциации.
Тандемная масс-спектрометрия: отклонения не обнаружены.
Установлен диагноз: нефротический синдром. Назначенное лечение: преднизолон 2 мг/кг/сут, коэнзим Q10 по поводу задержки психомоторного развития.
После 4 нед терапии преднизолоном протеинурия не уменьшилась. Проведена магнитно-резонансная томография головного мозга: обнаружены церебральная атрофия, двусторонняя гипомиелинизация в перивентрикулярном белом веществе, атрофия оптического нерва (рис. 1). При магнитно-резонансной спектроскопии метаболический пик достигнут не был. Учитывая отсутствие ответа на лечение, вместо преднизолона был назначен циклоспорин.
По результатам биопсии почки (11 почечных клубочков в материале) выявлено, что просветы почечных капилляров окклюзированы вследствие глобальной складчатости и коллапса базальных мембран с гипертрофией и гиперплазией прилежащих подоцитов и формированием псевдополулуний без воспалительных клеток и разрыва капсулы Боумена (рис. 1, А).

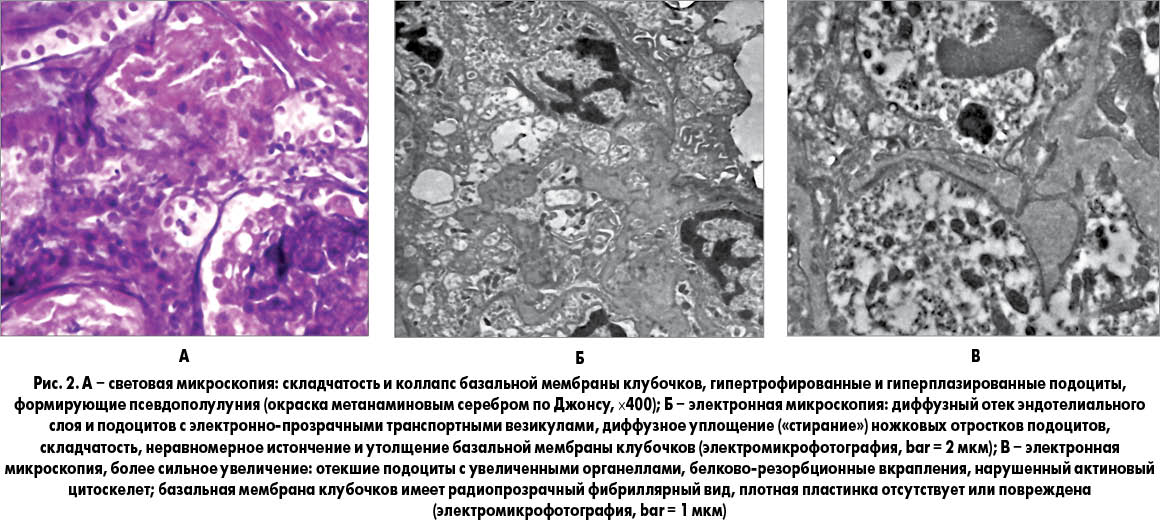
Гипертрофированные и гиперплазированные подоциты содержали в больших количествах интрацитоплазматические везикулы и белково-резорбционные вкрапления. Кроме того, наблюдались легкая дегенерация, регенерация и дилатация трубочек с легким и местами средневыраженным воспалением. Некоторые трубочки содержали красные резорбционные вкрапления. Иммунофлуоресцентный анализ показал отсутствие депозитов IgG, IgA, C3, C1q, фибрина, легких цепей иммуноглобулинов каппа и гамма. При электронной микроскопии полутонких срезов, окрашенных толуидином синим, выявлен коллапс базальных мембран клубочков с гипертрофией и гиперплазией подоцитов и формированием псевдополулуний. При окраске цитратом свинца и уранилацетатом обнаружены складчатость, неравномерное истончение и утолщение базальных мембран клубочков, сопровождавшиеся диффузным уплощением («стиранием»)«ножек» подоцитов с электронно-прозрачными транспортными везикулами, увеличенными органеллами и нарушенным актиновым цитоскелетом (рис. 2, Б). Базальные мембраны клубочков имели радиопрозрачный фибриллярный вид, при этом плотная пластинка, состоящая из коллагена IV типа и ламинина, во многих участках отсутствовала или была повреждена (рис. 2, В). Электронноплотные депозиты и тубероретикулярные включения в клубочках отсутствовали. В целом эти данные свидетельствовали в пользу диагноза коллапсирующей гломерулопатии.
Генетическое тестирование на мутацию подоцина патологии не выявило.
Клинического и лабораторного улучшения на фоне терапии циклоспорином не наблюдалось. Учитывая высокие сывороточные уровни мочевины (288 мг/дл) и креатинина (3,19 мг/дл), начат перитонеальный диализ. После 2 мес госпитализации был зафиксирован летальный исход вследствие тяжелой кандидозной септицемии, не ответившей на поддерживающее лечение и противогрибковую терапию.
Обсуждение
Синдром Галловея-Мовата (СГМ) – редкое аутосомно-рецессивное заболевание, характеризующееся сочетанием нефротического синдрома (НС) и поражением центральной нервной системы. С 1968 г., когда Галловей и Моват описали два случая раннего начала нефротического синдрома, микроцефалии и грыжи пищеводного отверстия у сиблингов, в мире было зарегистрировано более 60 пациентов с этим синдромом. СГМ – клинически гетерогенное заболевание, при этом наличие грыжи пищеводного отверстия более не является обязательным критерием диагноза.
Спектр поражения почек при СГМ может варьировать от легкой протеинурии без НС до стероидрезистентного НС с быстрым прогрессированием в терминальную стадию почечной недостаточности. НС обычно развивается в первые месяцы жизни (в среднем в возрасте 3 мес), хотя описано и более позднее его начало (в 44-198 мес). Раннее поражение почек (в возрасте до 3 мес) ассоциируется с более выраженными нарушениями развития головного мозга и ранним летальным исходом. Патологически поражение почек при СГМ включает ленточный фиброз и атрофию трубочек (как при токсичности ингибиторов кальциневрина), мезангиальную пролиферацию, микрокистозную дисплазию, диффузный мезангиальный склероз и фокальный сегментарный гломерулосклероз. Представленный клинический пример является третьим описанным случаем коллапсирующей гломерулопатии при СГМ.
Патогномоническими признаками СГМ при электронной микроскопии являются диффузное уплощение «ножек» подоцитов, неравномерное истончение базальной мембраны, атрофия трубочек и тубулоинтерстициальное воспаление. Все эти признаки присутствовали у данного пациента.
Неврологические аномалии наблюдаются у всех детей с СГМ и часто предшествуют почечным нарушениям. Ключевым морфологическим признаком заболевания является микроцефалия, которая может присутствовать уже при рождении или развиваться в более позднем возрасте. У некоторых пациентов микроцефалия сопровождается тяжелой задержкой развития и структурными аномалиями головного мозга, в частности мальформациями коры, гипомиелинизацией, нистагмом и мозжечковой атрофией. Более чем в половине случаев наблюдаются тяжелые, не отвечающие на лечение эпилептические припадки.
Дисморфизм при СГМ может проявляться такими признаками, как скошенный узкий лоб, низко поставленные большие подвижные уши, высокое нёбо, патологическая форма черепа, микрогнатия, арахнодактилия и грубые волосы.
Молекулярная основа развития СГМ на сегодня не известна. Нейроны и подоциты экспрессируют ряд общих белков, в частности синаптоподин, GLEEP1 и нефрин, однако у пациентов с СГМ мутации генов, отвечающих за синтез этих белков, отсутствовали. Анализ кандидатных генов, кодирующих белки базальной мембраны клубочков (LAMB2 и LAMB5) и белки подоцитов (ITGB1, ITGA3 и ACTN4), также не выявил отклонений. У 5 пациентов с СГМ (раннее начало НС, постнатальная микроцефалия, тяжелый интеллектуальный дефицит, мозжечковая атрофия) был идентифицирован дефицит гена WDR73. Этот ген, обнаруженный в мозговой и почечной ткани, кодирует синтез белка, содержащего повторы WD40, и играет ключевую роль в выживании клеток, а также в регуляции нейрональных и аксональных микротрубочек. В более поздних исследованиях было установлено, что мутация WDR73 часто встречается при нейродегенеративных заболеваниях с мозжечковой атрофией и не всегда сопровождается поражением почек. Кроме того, гломерулопатия, ассоциированная с мутацией WDR73, развивается в более позднем возрасте (в 5-12 лет). В приведенном клиническом случае мутация этого гена обнаружена не была. По-видимому, СГМ является генетически гетерогенным заболеванием, что и обусловливает неоднородную клиническую картину.
Cписок литературы находится в редакции.
Статья печатается в сокращении.
Zeybek С., Basbozkurt G., Hamcan S. et al. Collapsing Glomerulopathy in a Child with Galloway-Mowat Syndrome. Case Reports in Nephrology 2016; article 4386291.
Перевел с англ. Алексей Терещенко