


26 травня, 2020
Мукополісахаридоз 1 типу та хвороба Гоше: від теорії до практики
У рамках онлайн-вебінару, присвяченого орфанним захворюванням і організованого компанією Sanofi Genzyme, відбулася зустріч експертів, на якій детально розглянули патогенез, клінічні ознаки, діагностику й тактику лікування мукополісахаридозу 1 типу та хвороби Гоше.
Мукополісахаридоз 1 типу: як розпізнати пацієнта
 Професор кафедри педіатрії № 1 Національної медичної академії післядипломної освіти імені П.Л. Шупика, головний науковий співробітник відділення хвороб сполучної тканини у дітей з групою психосоматики та психотерапії ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», доктор медичних наук Олена Анатоліївна Ошлянська приділила увагу одному з орфанних захворювань – мукополісахаридозу (МПС) 1 типу (МПС І). Ця патологія належить до групи метаболічних захворювань сполучної тканини, пов’язаних із порушенням обміну глікозаміногліканів (ГАГ), що є наслідком мутації генів, які контролюють процес лізосомального гідролізу макромолекул.
Професор кафедри педіатрії № 1 Національної медичної академії післядипломної освіти імені П.Л. Шупика, головний науковий співробітник відділення хвороб сполучної тканини у дітей з групою психосоматики та психотерапії ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», доктор медичних наук Олена Анатоліївна Ошлянська приділила увагу одному з орфанних захворювань – мукополісахаридозу (МПС) 1 типу (МПС І). Ця патологія належить до групи метаболічних захворювань сполучної тканини, пов’язаних із порушенням обміну глікозаміногліканів (ГАГ), що є наслідком мутації генів, які контролюють процес лізосомального гідролізу макромолекул.
Основними спільними ознаками МПС є мультиорганність ураження, зміни рис обличчя та аномалії скелета. Додатковими ознаками є низький зріст, порушення функції серцево-судинної системи, дихання, гепатоспленомегалія та/або неврологічні розлади. Залучення всіх органів до патологічного процесу зумовлено наявністю сполучної тканини у кожній із систем організму (Marwan et аl., 2015).
До функцій сполучної тканини належать структуроутворювальна, трофічна, імунорегуляторна, репаративна та гомеостатична, тому патологічний процес при МПС характеризується вираженою дифузністю. Структурними компонентами сполучної тканини є клітини та міжклітинна речовина, що включає 3 типи волокон і гелеподібну основну речовину. Остання на 70% складається з води, неорганічних речовин і на 30% – із високомолекулярних сполук (глікозаміноглікани 7 видів: гіалуронова кислота, хондроїтин-4-сульфат, хондроїтин-6-сульфат, дерматансульфат, кератансульфати, гепарансульфати, гепарин).
Регулювання метаболізму сполучної тканини відбувається за участю 11 лізосомальних ферментів катаболізму ГАГ, гормонів (тироксин посилює деполімеризацію гіалуронової кислоти, глюкокортикоїди гальмують синтез і стимулюють розпад колагену та ГАГ, мінералокортикоїди мають протилежну дію), вітамінів С, групи В та D.
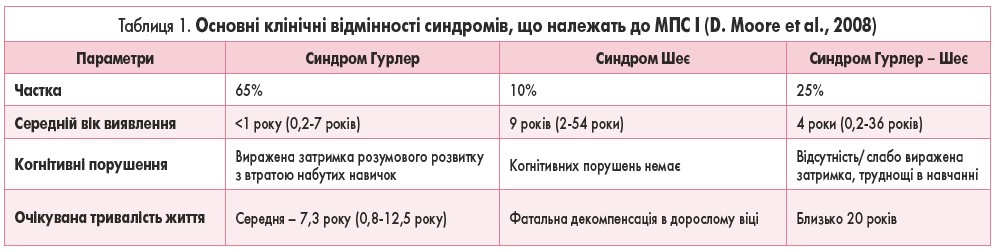
МПС І вперше описала у 1919 р. німецький педіатр Гертруда Гурлер, а у 1962 р. американський офтальмолог Гарольд Шеє описав пацієнта із синдромом, який назвали іменем дослідника. Частота виявлення синдрому Гурлер – 1:100 000, синдрому Шеє – 1:500 000, але у популяції ірландських мандрівників цей показник становить 1:371, а поширеність мутантного гена – 1:10 (P.J. Meikle et al., 1999; H.Y. Lin et al., 2009; G. Malm et al., 2008; D. Moore et al., 2008; A.M. Murphy et al., 2009).
Патофізіологія МПС І полягає у дефекті гена α-L-ідуронідази – IDUA 4p16.3, внаслідок чого активність вказаного ензиму знижується до ≤1%, порушується деградація ГАГ у лізосомах і прогресивно мультисистемно накопичується дерматан- та гепарансульфат в органах, де в нормі ці ГАГ виконують свої функції. Хоча варіанти МПС І є наслідком мутації в одному гені – IDUA 4p16.3, однак ступінь пенетрантності гена впливає на фенотип; основні клінічні відмінності синдромів Гурлер, Шеє та проміжної форми – Гурлер – Шеє представлені в таблиці.
При МПС І уражаються різні органи та системи:
1) дихальна система (S. Palmucci et al., 2013):
- гіпертрофія слизової оболонки, звуження дихальних шляхів;
- гучне дихання, зміни голосу, обструкції та рецидивні інфекції;
- апное уві сні, як наслідок – гіпоксія мозку і погіршення неврологічної симптоматики;
- кіфоз, зменшення рухливості грудної клітки;
- інфільтрація інтерстиційної тканини легень і рестриктивні порушення. Розлади функції дихальної системи є основною причиною смерті;
2) серцево-судинна система (V. Fesslova et al., 2009):
- гіпертрофічна кардіоміопатія;
- потовщення мітрального й аортального клапанів (у 89-100% пацієнтів), звуження судин, зменшення систолічного викиду;
- артеріальна гіпертензія;
- ішемічна хвороба серця;
3) нервова система (S. Palmucci et al., 2013):
- потовщення оболонок або нестабільність атланто-аксіального суглоба призводить до компресії спинного мозку та судин (може супроводжуватися порушенням ходи, дисфункцією сечового міхура, незграбністю рухів, судомами);
- аномалії головного мозку (підтверджені за допомогою магнітно-резонансної томографії), гідроцефалія;
- нейросенсорні порушення;
- тунельні синдроми (наприклад, синдром карпального каналу), позитивний симптом Тінеля, порушення чутливості перших трьох пальців;
4) опорно-рухова система:
- сповільнення росту після 1-2 років (ріст варіює в межах 100-165 см) на фоні правильної тілобудови (L.E. Polgreen et al., 2010);
- прогресуюча тугорухливість, сповільнення моторних навичок;
- обмеження відкривання рота;
- контрактури суглобів, зокрема дистальних міжфалангових суглобів кисті;
- симптом пташиної лапи та спускового гачка;
- кіфоз, сколіоз, гіперлордоз;
- дисплазія кульшового суглоба;
- вальгусна деформація кінцівок, варусна деформація стопи.
До рентгенологічних проявів ураження опорно-рухової системи належать:
- множинний дизостоз;
- аномалії хребців (гачкоподібні, гіперплазія передньої частини – горб, кіфоз) та ребер, вкорочені ключиці;
- збільшений череп, сплощення турецького сідла;
- потовщення діафізів, гіпоплазія епіфізів, точкові п’ясткові кістки;
- зменшення головок стегон, coxa valga, затримка осифікації вертлюгової ямки, дислокація головки стегнової кістки (R. Cimaz et al., 2009);
5) орган зору:
- відкладення субстрату в рогівці, що виявляється за допомогою щілинної лампи;
- помутніння рогівки або мегакорнеа, що супроводжуються світлобоязню та зниженням гостроти зору;
- вторинна глаукома розвивається внаслідок трабекулярної інфільтрації;
- дегенеративна ретинопатія й атрофія зорового нерва (G.A. Colville et al., 1996; E.F. Neufeld et al., 2001);
6) інше:
- утворення гриж черевної стінки (у 70% пацієнтів), що потребує хірургічного лікування;
- відсутність патології гіпофіза, щитоподібної залози, яєчок та яєчників.
Синдром Шеє є найлегшою формою МПС І, що зумовлено накопиченням лише дерматансульфату. У всіх пацієнтів із синдром Шеє захворювання маніфестує переважно ураженням опорно-рухового апарату (скутість у суглобах наявна у 44%), синдромом карпального каналу, можливе помутніння рогівки (у 28%), часті отоларингологічні проблеми (у 20%), пупкова грижа (у 16%). Згодом приєднуються класичні ознаки – у 66% пацієнтів наявні грубі риси обличчя, у 55% – гепатомегалія, у 52% – порушення слуху та у 34% – мовлення (R. Cimaz et al., 2006; S. Vijay et al., 2005).
При диференціації суглобових порушень від ювенільного артриту рекомендовано враховувати відсутність при МПС І:
- ознак локального запалення в ділянці суглобів (dolor, color, tumor, rubber et functio laesa);
- лабораторних ознак запалення;
- відповіді на терапію кортикостероїдами та нестероїдними протизапальними препаратами;
- радіографічних ознак ерозивного ушкодження кісток (S. Palmucci et al., 2013; R. Cimaz et al., 2009).
За умови виявлення у пацієнта ≥2 фенотипових ознак МПС І подальша тактика педіатра включає дотримання такого алгоритму:
1. Визначення екскреції ГАГ із сечею та типу ГАГ за допомогою тонкошарової хроматографії або електрофорезу, наприклад, у ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України» або Національній дитячій спеціалізованій лікарні «ОХМАТДИТ» МОЗ України (м. Київ).
Як правило, виявляють підвищення екскреції дерматан- і гепарансульфату, однак у дорослих із легким варіантом МПС результат може бути хибнонегативним.
2. Визначення активності лізосомальних ферментів у сухій краплі крові (Dried blood spot – DBS), лейкоцитах або фібробластах (зниження активності чи відсутність α-L-ідуронідази).
Для пацієнтів це дослідження є безкоштовним. Ферментна діагностика біоматеріалу проводиться в ARCHIMED Life Science GmbH Laboratories (Австрія). Діагностика здійснюється за рахунок ТОВ «Санофі-Авентіс Україна».
Більш надійним варіантом діагностики є дослідження лейкоцитів цільної крові або культивованих фібробластів. Можлива пренатальна діагностика на 8-10-му (біопсія хоріона) або 10-17-му тижні (амніоцентез).
3. Молекулярно-генетичне дослідження (мутація в гені IDUA 4p16.3 – остаточне підтвердження діагнозу МПС І; О. Gabrielli et al., 2009; 2016).
Лікування пацієнтів з МПС І передбачає два напрями:
1) патогенетичне лікування:
- ферментнозамісна терапія препаратом ларонідази – Альдуразим®* – з розрахунку 100 ОД/кг внутрішньовенно крапельно протягом 3-4 год 1 раз на тиждень;
- трансплантація гемопоетичних стовбурових клітин від 2 років (de Ru et al., 2011);
2) симптоматичне лікування при:
- гідроцефалії – шунтування у разі підвищення тиску ≥25-30 см вод. ст., або 18-22 мм рт. ст.;
- здавленні спинного мозку – декомпресійні операції до появи виражених порушень;
- порушенні функції кистей або нервової провідності за результатами електронейроміографії – декомпресія;
- артеріальній гіпертензії – корекція артеріального тиску;
- симптоматичній епілепсії – призначення протиепілептичних засобів у дозах, менших за середньотерапевтичні;
- офтальмологічних порушеннях – оперативне втручання;
- порушенні постави, тугорухомості суглобів, вальгусній деформації нижніх кінцівок ≥15° – ортопедична корекція;
- рецидивні інфекції – антибіотикотерапія, тонзилектомія, активна імунізація.
Важливо відмітити, що ферментнозамісна терапія препаратом Альдуразим® (за умови оптимального забезпечення симптоматичною терапією) успішно дозволяє впливати на (E.D. Kakkis et al., 2001; J.E. Wraith et al., 2007; L. Clarke et al., 2009; G. Petkovic et al., 2007; M. Sifuentes et al., 2007; M. Beck et al., 2007; R. Giugliani et al., 2008):
- щоденну активність (поліпшити рухливість, зменшити больовий синдром, нормалізувати сон);
- серцево-судинну систему (зменшити гіпертрофію лівого шлуночка та серцево-судинну недостатність, запобігти чи уповільнити ураження серця за умови збереження відносно нормальної функції серцевого м’яза);
- опорно-рухову систему (сприяє нормальному розвитку скелета, однак не у всіх пацієнтів препубертатного віку);
- дихальну систему (зменшити частоту епізодів апное та гіпопное, підвищити функціональну життєву ємність легень, відповідно до зросту збільшити об’єм легень);
- знизити виділення ГАГ із сечею (однак між екскрецією ГАГ та ефективністю терапії немає лінійної залежності);
- успішно перенести трансплантацію.
Професор О.А. Ошлянська на підтвердження терапевтичного ефекту препарату ларонідази навела клінічні випадки. Так, раннє призначення препарату хлопчику віком 5 місяців дало змогу уникнути розвитку типового для МПС І фенотипу та ураження органів, окрім незначного помутніння рогівки, безсимптомного ураження серця, скутості суглобів та мінімально вираженого множинного дизостозу. На противагу цьому у дівчинки-сибса, яка приймала ларонідазу з 5-річного віку, вже за наявності дещо грубих рис обличчя, скутості суглобів, помутніння рогівки, гепатоспленомегалії, мінімального множинного дизостозу, терапія зумовила регресування гепатоспленомегалії та стабілізацію патологічних клінічних ознак. Але й це, безумовно, розцінюється як позитивний результат (О. Gabrielli et al., 2016).
Спікер наголосила, що препарат ларонідази відповідно до наказу МОЗ України від 28.12.2019 № 2711 входить до номенклатури державних закупівель, тому безкоштовний для пацієнтів. МПС І входить до переліку рідкісних (орфанних) захворювань, які призводять до скорочення тривалості життя хворих або їx інвалідизації та для яких існують визнані методи лікування.
Єдиною перепоною на шляху до призупинення поліорганного ураження при МПС І є пізня діагностика захворювання, тому О.А. Ошлянська закликала педіатрів до прицільного пошуку об’єктивних ознак МПС (особливо ураження опорно-рухового апарату) у кожного потенційного хворого. Додатковим стимулюючим фактором є найбільша ефективність ферментнозамісної терапії у пацієнтів з легким, майже безсимптомним перебігом захворювання. При підозрі на МПС І варто провести визначення активності α-L-ідуронідази у сухій краплі крові (DBS), що є безкоштовним завдяки ТОВ «Санофі-Авентіс Україна».
Хвороба Гоше: діагностика та лікування
 Лікар-педіатр Центру орфанних захворювань Національної дитячої спеціалізованої лікарні «ОХМАТДИТ» МОЗ України Яна Ігорівна Дороніна представила інформацію про хворобу Гоше. Вона входить до групи лізосомальних хвороб накопичення з аутосомно-рецесивним типом успадкування, а саме сфінголіпідозів, тому в основі патогенезу захворювання лежить порушення обміну ліпідів. Спектр фенотипних проявів варіює від безсимптомного перебігу в осіб, що помирають у похилому віці, до несумісних із життям на ранніх етапах розвитку дитини.
Лікар-педіатр Центру орфанних захворювань Національної дитячої спеціалізованої лікарні «ОХМАТДИТ» МОЗ України Яна Ігорівна Дороніна представила інформацію про хворобу Гоше. Вона входить до групи лізосомальних хвороб накопичення з аутосомно-рецесивним типом успадкування, а саме сфінголіпідозів, тому в основі патогенезу захворювання лежить порушення обміну ліпідів. Спектр фенотипних проявів варіює від безсимптомного перебігу в осіб, що помирають у похилому віці, до несумісних із життям на ранніх етапах розвитку дитини.
Хвороба Гоше є хронічною прогресуючою гетерогенною мультисистемною патологією, що зустрічається з частотою 1:40 000-1:60 000, а в популяції євреїв-ашкеназі – 1:1 200. В основі патогенезу хвороби Гоше лежить генетично детерміноване зниження активності ферменту β-глюкоцереброзидази, що призводить до накопичення глюкоцереброзиду. У нормі метаболічний продукт мембран еритроцитів – глюкоцереброзид – поглинається макрофагами та розщеплюється під дією згаданого лізосомального ферменту на церамід і глюкозу (J. Stirnemann et al., 2017). При хворобі Гоше цей ензимозалежний процес є неможливим або неповноцінним, що призводить до накопичення глюкоцереброзиду в лізосомах макрофагів із подальшим збільшенням кількості патологічного включення та, відповідно, збільшенням об’єму макрофага (клітина Гоше). Оскільки макрофаги належать до ретикуло-ендотеліальної системи, то органами-мішенями при хворобі Гоше є печінка, селезінка, кісткова (включаючи кістковий мозок) і легенева тканина.
Виділяють кілька клінічних варіантів хвороби Гоше (G.M. Pastores et al., 2018):
- Хвороба Гоше 1 типу (ненейропатична форма) – зустрічається у 66-95% пацієнтів, є найбільш доброякісним, дебют хвороби можливий у будь-якому віці.
- Хвороба Гоше 2 типу (гостра нейропатична форма) – характеризується найбільш раннім дебютом і швидким перебігом.
- Хвороба Гоше 3 типу (хронічна нейропатична форма) – варіант, при якому клінічні ознаки з’являються у віці 5-6 років, а перебіг є повільнопрогресуючим.
- Перинатально-летальна форма.
- Серцево-судинна форма.
Ранні симптоми хвороби Гоше, як правило, є відображенням гематологічних проявів. Хвороба може маніфестувати такими ознаками:
- спленомегалія (у 90-100% пацієнтів) – збільшення селезінки у 2-80 разів від початкового розміру, що призводить до абдомінального дискомфорту; інфаркти та фіброз селезінки, як наслідок – портальна гіпертензія;
- тромбоцитопенія (у 90-100% пацієнтів) через гіперспленізм та порушення функції тромбоцитів, наслідок – кровотеча з носа/ясен (у 90%), петехії, екхімози;
- анемія (у 90% хворих) внаслідок гіперспленізму, у зв’язку з чим пацієнт може скаржитися на хронічну втому;
- гепатомегалія (у 70% пацієнтів) і порушення функції печінки;
- лейкопенія (у 40% пацієнтів), відповідно – схильність до розвитку інфекцій, вищий ризик розвитку сепсису;
- гіперферитинемія (у 20% хворих);
- кісткові порушення: біль у кістках, кісткові кризи, васкулярний некроз, кісткові деформації, остеонекроз, переломи (Belgian Working Group on Gaucher disease, 2016).
Що стосується найпоширеніших прогресуючих і незворотних уражень скелета при хворобі Гоше, то у пацієнтів закономірно розвиваються такі патологічні процеси:
- інфільтрація кісткового мозку, внаслідок чого поступово виникає ремоделювання кістки, деформація кісток у вигляді колб Ерленмеєра;
- хронічна ішемія кісток призводить до кісткових інфарктів та остеонекрозу (локалізована загибель кістки) головки та шийки стегнової кістки, проксимального відділу плечової кістки, великогомілкової кістки та хребців;
- розвиток літичних уражень – невеликі ерозії на кістках за рахунок локалізованих ділянок руйнування;
- патологічні переломи, руйнування кісток і суглобів, деформація, колапс тіл хребців, що супроводжуються відростанням периферичних клітин, «провисанням», формуванням Н-подібної форми;
- остеосклероз – ненормальне затвердіння або збільшення щільності кістки за рахунок гіперактивного запалення у ній (Belgian Working Group on Gaucher disease, 2016).
Клінічними ознаками розвитку патологічного процесу в кістковій тканині є біль (гострий/хронічний, епізодичний/постійний, неспецифічний, ниючий) і кісткові кризи (сильний біль, лихоманка, лейкоцитоз, зниження рухливості), що часто потребують застосування наркотичних препаратів з метою знеболювання. Біль імітує гострий остеомієліт, може супроводжувати кістковий інфаркт, що розвивається паралельно (Belgian Working Group on Gaucher disease, 2016).
Я.І. Дороніна акцентувала увагу на незворотності ураження кісткової тканини у пацієнтів з хворобою Гоше, саме тому вчасна діагностика, лікування та профілактичні заходи є ключем для уникнення серйозних наслідків ураження кісток.
Хвороба Гоше 2 типу (гостра нейропатична форма) має інший фенотип і клінічні ознаки. При цьому типі тривалість життя складає в середньому 3-6 міс, а на перший план виходять неврологічні прояви, органомегалія та гематологічні порушення, зокрема страбізм, над’ядерний параліч погляду, порушення дихання, зниження смоктального та ковтального рефлексів, дистонія, епілепсія, тризм, перерозгинання шиї.
Хвороба Гоше 3 типу (хронічна нейропатична форма) є більш сприятливою в плані тривалості життя – смерть настає до 2 років. Але за умови вчасної діагностики та початку ферментнозамісної терапії тривалість життя подовжується до дорослого віку. Клінічними особливостями хвороби Гоше 3 типу є комбінація фенотипу хвороби Гоше 1 типу та неврологічних порушень. Залежно від фенотипових ознак виділяють три варіанти хвороби Гоше 3 типу:
- тип 3а (Норботтен-тип): прогресуюча деменція, атаксія, міоклонус, горизонтальний супрануклеарний парез погляду;
- тип 3b: виражені вісцеральні та кісткові порушення, супрануклеарний парез погляду;
- тип 3с: супрануклеарний парез погляду, помутніння рогівки, серцево-судинні кальцифікати.
При виявленні типових ознак хвороби Гоше алгоритм діагностики включає проведення більш прицільного клінічного обстеження, а також морфологічне, біохімічне та молекулярно-генетичне дослідження. Морфологічне дослідження спинного мозку не є обов’язковим, враховуючи інвазивність процедури та високу частоту хибнонегативних результатів (у пунктаті 35% усіх пацієнтів з хворобою Гоше були виявлені клітини Гоше). Тому цей метод використовується з метою диференційної діагностики від інших лімфопроліферативних захворювань (J. Stirnemann et al., 2017).
Лабораторні дослідження дозволяють виявити панцитопенію, гіперферитинемію, гіпохолестеринемію, скорочення протромбінового часу.
Додатково можуть бути застосовані інструментальні допоміжні методи діагностики:
- ультразвукова та магнітно-резонансна томографія (МРТ) печінки й селезінки (вогнищеве ураження органів, динаміка об’єму органів, що можна використати для контролю ефективності ферментнозамісної терапії);
- денситометрія та МРТ кісток (найчутливіші методи діагностики остеопенії та інфільтрації кісткового мозку);
- МРТ головного мозку, електроенцефалографія та нейропсихометрія (додатково до нейроофтальмологічних тестів, аналізу периферичного слуху, дослідження функції нервової системи та руху очей);
- доплер-ехокардіографія (у пацієнтів після спленектомії).
Специфічна діагностика хвороби Гоше 1 типу здійснюється за таким алгоритмом:
- Простим, малоінвазивним і доступним скринінговим методом діагностики є метод сухої краплі крові. Важливо, що зібраний матеріал можна транспортувати. Ферментна діагностика біоматеріалу здійснюється за рахунок ТОВ «Санофі-Авентіс Україна» в ARCHIMED Life Science GmbH Laboratories (Австрія).
- Для верифікації діагнозу пацієнта направляють до лабораторії Центру орфанних захворювань Національної дитячої спеціалізованої лікарні «ОХМАТДИТ» для визначення активності ферменту β-глюкоцереброзидази в лейкоцитах крові, яке є золотим стандартом діагностики хвороби Гоше.
- Після виявлення зниження активності ферменту за протреби проводять молекулярно-генетичне дослідження.
- Додатковим специфічним методом дослідження є визначення біомаркерів, що корелюють зі ступенем активності хвороби Гоше, а саме активності ферментів хітотріозидази, CCL18, тартратрезистентної кислої фосфатази та ангіотензинперетворювального ферменту (Bodamer et al., 2010).
Оскільки хвороба Гоше належить до орфанних захворювань і включена у перелік нозологій, лікування яких є безкоштовним для пацієнта, призначення замісної ферментної терапії здійснюється на базі Центру орфанних захворювань Національної дитячої спеціалізованої лікарні «ОХМАТДИТ» в порядку, установленому згідно з наказами МОЗ України від 19.10.2015 № 683 та від 05.02.2015 № 50.
Пожиттєва безперервна ферментнозамісна терапія, що є золотим стандартом лікування хвороби Гоше 1 та 3 типів, більше ніж 20 років представлена іміглюцеразою (Церезим® 400 ОД**), також при хворобі Гоше 1 типу в Україні використовують велаглюцеразу альфа і таліглюцеразу альфа.
Дозу іміглюцерази підбирають індивідуально, відповідно до клінічного статусу пацієнта та його молекулярних показників. Протягом усього часу замісної ферментної терапії через кожні 6, 12 або 24 місяці проводиться клінічний, лабораторний та інструментальний моніторинг.
Підтверджено, що лікування препаратом Церезим® 400 ОД призводить до зменшення гепатомегалії на 30-40% та спленомегалії на 50-60% (N.J. Weinreb et al., 2002). Протягом 4 років лікування як мінімум 70% пацієнтів успішно досягають мети терапії (N.J. Weinreb et al., 2008). Додатковими показниками ефективності препарату Церезим® 400 ОД є (N. Weinreb et al., 2007; Andersson et al., 2008; Charrow et al., 2007):
- збільшення кількості тромбоцитів, рівня гемоглобіну, усунення залежності від гемотрансфузій;
- запобігання розвитку гепатоспленомегалії, портальної та легеневої гіпертензії;
- зменшення кісткових кризів і болю у кістках;
- відновлення нормального росту у дітей;
- досягнення оптимальної пікової кісткової маси у дітей;
- запобігання остеонекрозу, остеомієліту та інших кісткових ускладнень (за умови вчасного початку терапії);
- підвищення мінеральної щільності кісток;
- підвищення якості життя;
- покращення фізичного та психічного здоров’я пацієнтів з хворобою Гоше.
Альтернативою ферментнозамісній терапії можуть бути пероральна субстрат-редукуюча терапія та терапія фармакологічними шаперонами, остання наразі досліджується.
Комплексний підхід до ведення пацієнтів з хворобою Гоше полягає у розгляді доцільності часткової або тотальної спленектомії; трансфузії еритроцитарної або тромбоцитарної маси, свіжозамороженої плазми крові; призначення аналгетиків при болю в кістках (однак слід уникати застосування нестероїдних протизапальних засобів при помірній і тяжкій тромбоцитопенії); оперативного ортопедичного втручання (оcтеосинтез, ендопротезування суглобів, артродезуючі операції, коригуюча остеотомія, некрсеквестректомія); призначення вітамінів, мікроелементів (Ca2+, вітамін D3; P. Mistry et al., 2007).
На завершення доповіді Я.І. Дороніна представила клінічний випадок хвороби Гоше у дівчинки 6 років. У дитини спостерігали збільшення об’єму живота, рецидивуючий геморагічний висип, носові кровотечі, затримку росту та збільшення маси тіла, біль у нижніх кінцівках, часті епізоди гострих респіраторних захворювань. При обстеженні було виявлено залізодефіцитну анемію, гіперферитинемію, панцитопенію, коагулопатію, підвищення рівня лужної фосфатази, гепатоспленомегалію, початкові ознаки портальної гіпертензії, остеопенію легкого ступеня за даними подвійної енергетичної рентгенівської абсорбціометрії, розширення дистальних відділів трубчастих кісток за типом колб Ерленмеєра, початкові ознаки синовіїту. Діагноз було підтверджено на основі зниження активності β-глюкозидази до 2,6 нмоль/год/мл, підвищення активності хітотріозидази до 7293 нмоль/год/мл та остаточно – за допомогою генетично-молекулярного аналізу.
У цьому клінічному випадку призначення іміглюцерази у дозі 30 ОД/кг 1 раз на 2 тижні внутрішньовенно крапельно у віці 7 років сприяло регресуванню гепатоспленомегалії, нормалізації фізичного розвитку, показників гемограми, мінеральної щільності кісткової тканини.
Таким чином, у клінічній практиці однією з найсерйозніших проблем при хворобі Гоше залишається її раннє виявлення. Своєчасна діагностика та призначення патогенетичної терапії забезпечують усунення проявів інвалідизуючих симптомів хвороби, що може сприяти повній реабілітації пацієнтів із цією тяжкою недугою (К. Wenstrup et al., 2007; J. Charrow et al., 2007).
*Лікарський засіб Альдуразим®, концентрат для розчину для інфузій, 100 ОД/мл, № 1, зареєстрований в Україні. Р.П. UA/8093/01/01. Наказ МОЗ № 1449 від 03.08.2018. Зміни внесено наказом МОЗ № 2205 від 31.10.2019.
** Лікарський засіб Церезим® 400 ОД, порошок для приготування концентрату для розчину для інфузій по 400 ОД, зареєстрований в Україні. Р.П. № UA/8659/01/02. Наказ МОЗ № 1504 від 16.08.2018. Зміни внесено наказом МОЗ № 2205 від 31.10.2019.
Підготувала Маргарита Марчук
Тематичний номер «Педіатрія» №2 (53) 2020 р.