


30 червня, 2020
Поширеність хвороби Помпе у пацієнтів із підвищенням активності креатинкінази та слабкістю м’язів кінцівок
Хвороба Помпе (ХП), або хвороба накопичення глікогену 2-го типу – це мультисистемне метаболічне захворювання, спричинене недостатністю ферменту лізосомної кислої α‑1,4-глюкозидази (GAA). Z. Lukacs et al. провели проспективний скринінг великої вибірки пацієнтів із підвищенням активності сироваткової креатинкінази (КК) та/або слабкістю м’язів кінцівок (дистрофією Лейдена) на предмет недостатності GAA за допомогою дослідження сухої краплі крові (DBS). Отримані результати опубліковані у виданні Neurology (2016; 87 (3): 295‑298).
ХП із пізнім початком характеризується різними віком дебюту захворювання та фенотипом. У більшості хворих основними клінічними проявами є аксіальна і проксимальна слабкість скелетних м’язів. Однак захворюваність та смертність пацієнтів, зокрема, пов’язані з дихальною недостатністю (Güngör et al., 2011). ХП із пізнім початком проявляється широким спектром симптомів, серед яких окремі нервово-м’язові симптоми на первинному етапі, через що хворобу можна не діагностувати (Schüller et al., 2012).Затримання у встановленні діагнозу зазвичай складає від 7 до 10 років (Kishnani et al., 2013).
Можливо, настання епохи ферментозамісної терапії вплинуло на необхідність ліпшої обізнаності щодо ХП. Тож Z. Lukacs et al. виконали проспективний скринінг пацієнтів із підвищенням активності сироваткової КК та/або слабкістю м’язів кінцівок (дистрофією Лейдена) на предмет недостатності GAA шляхом дослідження DBS.
Матеріали й методи дослідження
Для включення у дослідження пацієнтів відбирали шляхом медичного обстеження, яке проводили у семи стаціонарних та амбулаторних клініках для лікування нервово-м’язових захворювань Німеччини й Великої Британії упродовж 2009‑2014 рр. В усіх хворих було отримано усну та письмову згоду на участь. Дослідження було виконане відповідно до Гельсинської декларації та ухвалене етичною комісією Університету Людвіга Максиміліана у Мюнхені (Німеччина). Для описового статистичного аналізу використовували програму для Windows SPSS (версія 23.0; IBM, Нью-Йорк, США).
Критерії включення
Для участі у дослідженні було відібрано 3076 пацієнтів віком від 18 до 89 років (медіана – 48 років; 58,2% – жінки). Вибірка складалася з осіб із некласифікованою проксимальною та/або аксіальною слабкістю м’язів кінцівок (дистрофією Лейдена) та/або стійким нез’ясованим підвищенням рівня сироваткової КК. Під дистрофією Лейдена малася на увазі слабкість проксимальних м’язів рук та ніг, зокрема плечового пояса, плеча, аксіальної паравертебральної, тазової області та стегон (Güngör et al., 2011; Schüller et al., 2012). Неодноразове збільшення вмісту КК як критерію включення було визначене як перевищення верхньої границі норми принаймні вдвічі: <180 МО/л для жінок та <200 МО/л для чоловіків.
Взяття зразків, флуорометрія та генетичне секвенування
Зразки DBS забирали з периферичної вени за допомогою пробірки, покритої етилендиамінтетраоцтовою кислотою. Далі кров було негайно нанесено на фільтраційний папір. Усі зразки анонімізували та досліджували в Лабораторії обміну речовин Інституту клінічної хімії Університетського медичного центру м. Гамбург (Німеччина). Для дослідження був використаний метод флуорометричного аналізу на предмет недостатності GAA (Lukacs et al., 2010). Зразки DBS, в яких було виявлено зменшену ферментативну активність, перевіряли на мутації гена GAA.
Секвенування за Сенгером кодувального та фланкувального екзон регіону гена GAA проводили, коли активність GAA у DBS була <0,9 нмоль/пробу за 21 год у двох незалежно взятих зразках DBS.
Результати дослідження
Демографічні характеристики пацієнтів та клінічні прояви підсумовані в таблиці 1. Результати активності GAA 2844 осіб (92,2%) були в межах норми. Середній рівень КК у всіх учасників становив 563 МОд/л; нормальний діапазон значень – 15 тис. МОд/л. Активність GAA у DBS була нижчою за граничний показник 0,9 нмоль/пробу за 21 год у 232 хворих (середній вік – 40,7 року).
Активність GAA у DBS серед неуражених осіб при використанні інгібітора GAA акарбози переважно становила 1,73 нмоль/пробу (медіана – 1,22 нмоль/пробу) впродовж 21 год. За допомогою мутаційного аналізу в 42 жінок (56%) та 32 чоловіків було встановлено діагноз ХП із пізнім початком. Показники активності GAA у DBS при застосуванні акарбози у всіх пацієнтів із підтвердженою ХП склали 0,18 нмоль/пробу (медіана – 0,12 нмоль/пробу) протягом 21 год. У 70 хворих секвенування гена GAA виявило дві гетерозиготні відомі патогенні мутації, зокрема за частою мутацією сайту сплайсингу гена GAA c.-32‑13T>G на одному з алелів, що своєю чергою підтвердило діагноз.
Також за граничною ферментативною активністю GAA було ідентифіковано 158 можливих носіїв ХП. Повторне дослідження DBS показало нормальні результати у 123 із них. Альтернативний діагноз було остаточно поставлено 52 із 123 учасників, а саме рецесивну кінцівково-поясну дистрофію (n=32), м’язову дистрофію Бекера (n=4), міотонічну дистрофію 2-го типу (n=8), міозит (n=6), кінцівково-поясний міастенічний синдром (n=1), атрофію м’язів спини 3-го типу (n=1). Діагноз 95 осіб із граничними значеннями ферментативної активності GAA залишився невстановленим. У 24 із цих пацієнтів було неможливо провести повторне дослідження DBS; 11 виявилися гетерозиготними за частою мутацією сайту сплайсингу гена GAA c.-32‑13T>G, при цьому мутацію другого гена GAA не встановили.
Обговорення
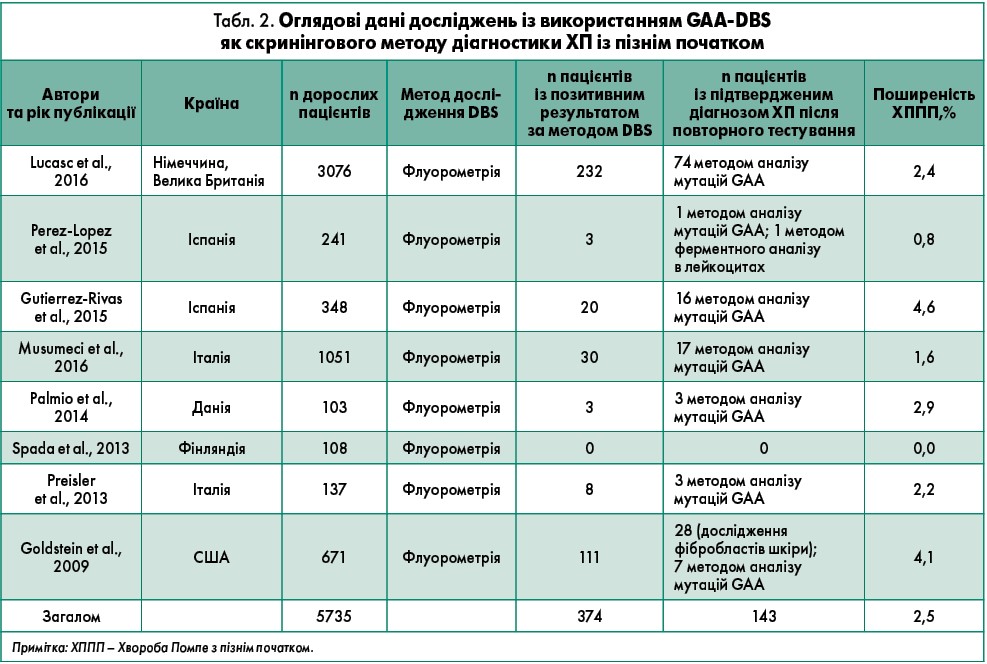
Наразі це найбільш проспективне когортне скринінгове дослідження хворих на ХП із пізнім початком та некласифіковані м’язові хвороби. Ефективність методу GAA було описано при вивченні вибірки немовлят та в межах інших фокусованих досліджень ХП (табл. 2) (Gutiérrez-Rivas et al., 2015; Musumeci et al., 2016; Palmio et al., 2014; Spada et al., 2013; Preisler et al., 2013; Goldstein et al., 2009; Perez-Lopez et al., 2015; Schoser, Toscano, 2013). Усі 74 пацієнти, яким було вперше поставлено діагноз ХП, мали ключові клінічні прояви хвороби з пізнім початком. Важливим наслідком дослідження є те, що у 2,4% дорослих хворих із некласифікованою дистрофією Лейдена та/або підвищенням рівня КК було встановлено ХП. Ці дані відповідають результатам менших випробувань, проте з комбінованою поширеністю хвороби у 2,5% (Gutiérrez-Rivas et al., 2015; Musumeci et al., 2016; Palmio et al., 2014; Spada et al., 2013; Preisler et al., 2013; Goldstein et al., 2009; Perez-Lopez et al., 2015). Хоча були використані однакові методи флуорометрії, всі вісім досліджень DBS показали невеликі відмінності у поширенні ХП. Ці розбіжності можуть відображати різницю в частоті захворюваності на рідкісну хворобу залежно від країни (van der Ploeg, Reuser, 2008; Kishnani et al., 2013).
Також недостатність активності GAA не завжди можна підтвердити методом DBS у повторно взятому зразку. Серед відомих причин є взяття крові після переливання, неправильне взяття та нанесення DBS, а також зовнішні фактори, як-то висока температура під час транспортування. Навіть у цьому дослідженні, при залученні досвідчених центрів, було неможливо уникнути помилок при нанесенні крові або щодо дотримання умов зовнішнього середовища.
Варто зазначити, що був проведений внутрішній контроль усіх проб DBS шляхом вимірювання активності нейтральної мальтази, який застосовували щодо першого зразка досліджених пацієнтів. Однак лишається ймовірність псевдопозитивного результату. Тому автори рекомендують підтверджувати чи спростовувати отримані дані за допомогою інших тканин, наприклад фібробластів / м’язової тканини чи золотого стандарту, коли це стосується GAA, – генетичного секвенування за Сенгером. Класично при ХП із пізнім початком за проведення біопсії м’язової тканини можна побачити автофагічні вакуольні міопатичні зміни, накопичення глікогену, а також включення, позитивні на кислу фосфатазу. Останнє може спостерігатися без очевидного накопичення глікогену. Однак фахівцям добре відомо, що накопичення глікогену та включення кислої фосфатази бувають відсутні, але це не виключає проведення DBS. Можливо, дослідження методом DBS варто виконувати до взяття біопсії, якщо у клініциста є підозра щодо розвитку в пацієнта ХП.
Враховуючи відносно рідкісні випадки ХП із пізнім початком, неспецифічні клінічні ознаки проксимальної слабкості та підвищення рівня КК, а також зазвичай відсутність ознак порушень при проведенні біопсії м’язової тканини, навіть досвідчені спеціалісти з нервово-м’язових захворювань можуть не запідозрити даний діагноз. Тож автори дійшли висновку, що доцільно проводити дослідження методом DBS на ранніх етапах діагностики пацієнтів із некласифікованою дистрофією Лейдена чи наявним підвищенням активності КК для виключення діагнозу ХП із пізнім початком. Ключовими клінічними ознаками, що мають насторожити лікаря та спонукати провести скринінг за допомогою DBS на предмет ХП із пізнім початком, є ураження м’язів шиї, таза, дихальної мускулатури та персистувальне зростання рівня КК.
Висновки
З’являється все більше доказів на підтвердження того, що на ранніх етапах у хворих на ХП із пізнім початком доцільно застосовувати ферментозамісну терапію. Її варто призначати, перш ніж відбудеться значне, можливо, незворотне пошкодження м’язової тканини. Таке лікування є значно ефективнішим порівняно з тим, що розпочате на більш пізніх стадіях. Це ще раз наголошує на важливості ранньої діагностики (van der Ploeg, Reuser, 2008; Musumeci et al., 2016; Goldstein et al., 2009; Schoser, Toscano, 2013).
Довідка «ЗУ»
В Україні карти для забору біоматеріалу (DBS) та лабораторну діагностику надає ТОВ «Санофі-Авентіс Україна». Для пацієнтів обстеження абсолютно безкоштовне. Ферментна діагностика проводиться в метаболічній лабораторії ARCHIMED Life Science GmbH Laboratories (Австрія) або лабораторії Центру орфанних захворювань НДСЛ «ОХМАТДИТ» (м. Київ).
Підготувала Юлія Паламарчук
Тематичний номер «Неврологія, Психіатрія, Психотерапія» № 2 (53) 2020 р.