


25 листопада, 2016
Диагностика идиопатического легочного фиброза

 Идиопатический легочный фиброз (ИЛФ) не относится к категории редких заболеваний легких – заболеваемость ИЛФ сопоставима по уровню с заболеваемостью туберкулезом в странах Западной Европы. Вместе с тем практикующие пульмонологи и рентгенологи не владеют достаточным уровнем знаний о клинической и радиологической семиотике этого заболевания.
Идиопатический легочный фиброз (ИЛФ) не относится к категории редких заболеваний легких – заболеваемость ИЛФ сопоставима по уровню с заболеваемостью туберкулезом в странах Западной Европы. Вместе с тем практикующие пульмонологи и рентгенологи не владеют достаточным уровнем знаний о клинической и радиологической семиотике этого заболевания.
Идиопатический легочный фиброз – это специфическая форма хронической прогрессирующей интерстициальной фиброзирующей пневмонии неизвестной природы, наблюдаемая в основном у лиц пожилого и старческого возраста, ограниченная поражением легких и ассоциированная с гистопатологическим и/или радиологическим паттерном обычной интерстициальной пневмонии [1].
 ИЛФ характеризуется чрезвычайно неблагоприятным прогнозом – средняя продолжительность жизни больных от момента установления диагноза составляет от 2,5 до 3,5 лет.
ИЛФ характеризуется чрезвычайно неблагоприятным прогнозом – средняя продолжительность жизни больных от момента установления диагноза составляет от 2,5 до 3,5 лет.
За последние 15 лет было проведено 7 крупных исследований по изучению заболеваемости и распространенности ИЛФ с использованием как широких, так и узких критериев верификации диагноза. В исследовании E.R. Fernandez-Perez и соавт. [2], проведенном в США, применялись жесткие критерии учета больных – возраст >50 лет, наличие паттерна обычной интерстициальной пневмонии (ОИП) при патогистологическом изучении биоптатов легких или достоверных признаков ОИП-паттерна при компьютерной томографии (КТ). В результате установлено, что заболеваемость ИЛФ составила 8,8 случая на 100 тыс. населения в год, распространенность – 27,9 случая на 100 тыс.
Этиология заболевания до настоящего времени не установлена. Рассматриваются потенциальные факторы риска – курение, пыль металлов и древесины, гастроэзофагеальный рефлюкс, генетическая предрасположенность.
Предполагается, что вследствие воздействия какого-то неустановленного фактора повреждается нормальный структурный слой, который разделяет полость альвеолы от просвета капилляра. Он состоит из эпителиальных клеток, базальной мембраны и эндотелиальных клеток капилляра. Далее включаются репаративные процессы с высвобождением эпителиальноклеточных медиаторов и проникновением факторов крови, включая факторы коагуляции, в просвет альвеол. Процесс приобретает организующий характер, инициируется рубцевание, и на каком-то этапе процесс фиброзирования выходит из-под контроля. Причины повреждения, в равной мере как и причины прогрессирующего фиброзирования, до настоящего времени не установлены.
Клинические симптомы
ИЛФ обычно проявляется постепенно нарастающей одышкой, реже – непродуктивным кашлем, который нередко имеет приступообразный характер и отличается рефрактерностью к противокашлевым средствам. Отличительной особенностью ИЛФ является незаметное появление симптомов – пациент обычно затрудняется указать сроки появления одышки с точностью до полугода, а иногда и более. Среди больных преобладают мужчины. ИЛФ типично возникает в шестом или седьмом десятилетии жизни. Пациенты в возрасте до 50 лет встречаются редко, у таких больных впоследствии могут проявиться симптомы системного заболевания соединительной ткани, отсутствовавшие на момент установления диагноза ИЛФ. У большинства больных период от начала появления симптомов до обращения к врачу превышает 6 месяцев.
Деформация ногтевых фаланг в виде «барабанных палочек» отмечается у 25-50% пациентов. При аускультации феномен «треск целлофана» в конце выдоха определяется в нижних отделах, а затем над всей поверхностью легких.
Признаки хронического легочного сердца (периферические отеки) могут наблюдаться в поздних стадиях заболевания.
Клиническое течение ИЛФ характеризуется постепенным ухудшением состояния больных, однако нередко наступает резкое прогрессирование, связанное с вирусной инфекцией, развитием пневмонии или диффузного альвеолярного повреждения (ДАП).
Обязательным для диагностики ИЛФ является присутствие морфологического паттерна обычной интерстициальной пневмонии [1, 3].
Гистопатологический паттерн ОИП
Отличительным гистопатологическим признаком и главным диагностическим критерием является гетерогенное чередование при малом увеличении областей фиброза и сотовых изменений с участками менее пораженной или нормальной паренхимы (рис. 1) [1, 4].
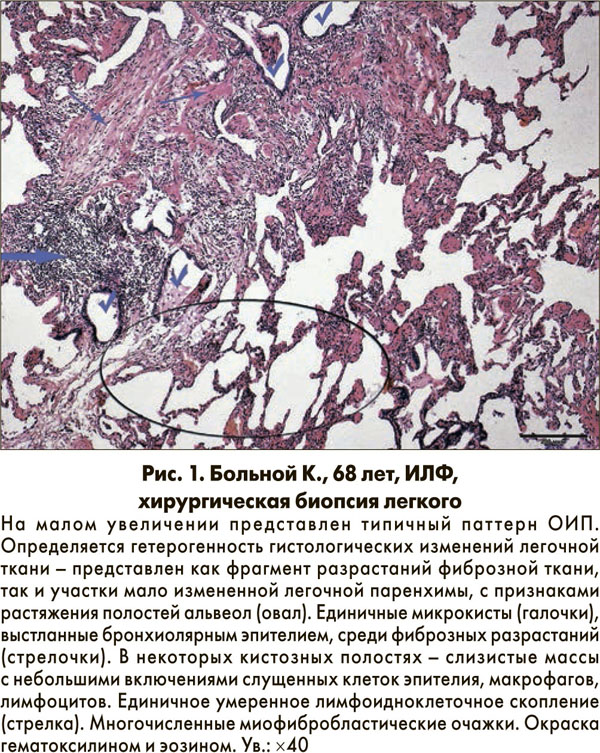
Эти гистопатологические изменения часто наблюдаются в субплевральных и парасептальных участках. Воспаление обычно слабое, характеризуется пятнистой интерстициальной инфильтрацией лимфоцитами и плазматическими клетками с гиперплазией пневмоцитов 2 типа и бронхиолярного эпителия. Зоны фиброза состоят преимущественно из плотного коллагена, хотя также постоянно находят рассеянные выпуклые субэпителиальные очаги пролиферирующих фибробластов и миофибробластов (так называемые очаги фибробластов) [5].
Фибробластические фокусы представляют собой мелкие очажки с наличием фибробластов и миофибробластов в нежно окрашенном межклеточном веществе (рис. 2).
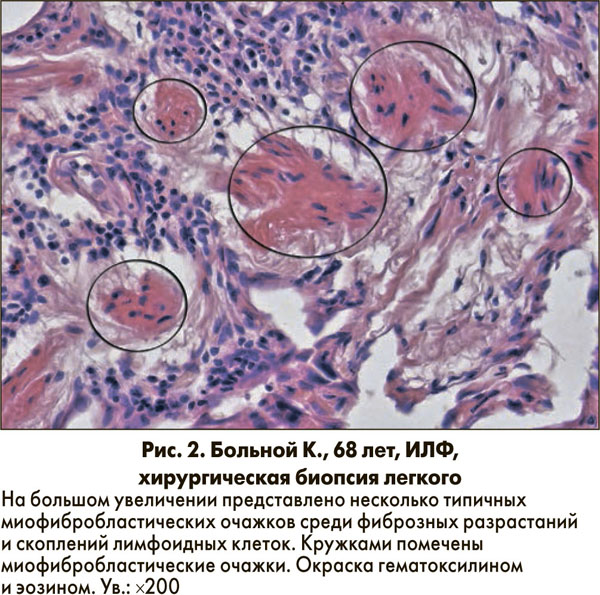
Фибробластические фокусы не являются специфичным признаком ОИП, однако их наличие, причем в значительном количестве, важно для установления диагноза.
«Сотовые» изменения обнаруживают в большинстве операционных биопсий легочной ткани, они представляют собой расширенные воздушные пространства, обычно выстланные кубическим эпителием, среди очагов фиброза (рис. 3) [3].
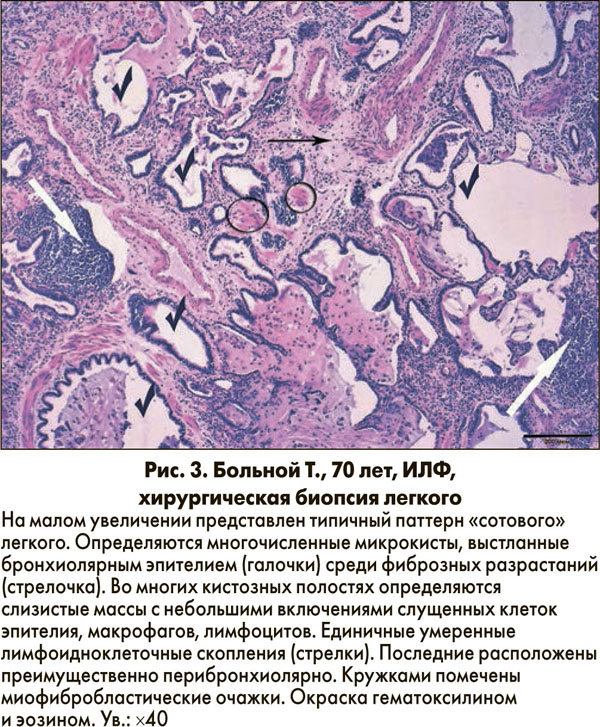
Внутри микрокист часто присутствуют слизистые массы, а также могут быть включения из альвеолоцитов, клеток крови и клеток воспаления [1].
Участки фиброза, не связанные с «сотами», – еще один типичный гистологический признак ОИП. В очагах фиброза и в стенках «сот» часто можно видеть гиперплазию гладких мышц, которая в части наблюдений может быть весьма выраженной.
При ОИП возможно обнаружение плоскоклеточной метаплазии бронхиолярного эпителия, а также эпителия, выстилающего аденоматозные «сотовые» структуры, редко – оссификации, облитерирующего эндартериита. Однако эти изменения неспецифичны и развиваются вторично.
При исследовании биопсий в фазу острой ОИП имеет место сочетание картины ОИП и ДАП или реже – организующей пневмонии.
ДАП при обострении ОИП характеризуется мозаичной картиной поражения легких, с утолщением межальвеолярных перегородок за счет фибробластов и миофибробластов с минимальной воспалительной инфильтрацией, выраженной гиперплазией альвеолоцитов II типа, иногда с признаками их атипии, появлением гиалиновых мембран, фибриновых тромбов в мелких сосудах, плоскоклеточной метаплазией бронхиолярного эпителия.
Паттерн ОИП не является патогномоничным для ИЛФ, он встречается и при других интерстициальных заболеваниях легких, в частности при диффузных заболеваниях соединительной ткани с поражением легких [6]. То есть ОИП-паттерн не обязательно указывает на наличие ИЛФ, но во всех случаях ИЛФ обязательно присутствие морфологических признаков ОИП [1].
В 2000 году Американское торакальное общество и Европейское респираторное общество опубликовали первое международное положение по диагностике и лечению ИЛФ – American Thoracic Society, European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement (2000) [3].
Положение предусматривало обязательную верификацию диагноза методом хирургической биопсии легких. Эта рекомендация была основана на результатах оценки достоверности диагностики ИЛФ на основе клинических симптомов, данных рентгенографии и КТ органов грудной полости в сравнении с результатами биопсии. Так, достоверность диагноза, основанного только на клинических данных, составила 62%, а применение рентгенографии и КТ повышало точность диагностики всего до 76%.
Опыт научных исследований, накопленный с момента опубликования в 2000 году соглашения по ИЛФ, обусловил необходимость уточнения критериев диагностики заболевания и пересмотра некоторых принципов лечения больных. Прежде всего, это было связано с бурным развитием технологий в области КТ. За следующие 10 лет диагностические возможности КТ возросли настолько, что по результату морфологической диагностики этот метод стал успешно конкурировать с методом патогистологического исследования.
В связи с этим в 2011 году было опубликовано новое руководство по диагностике и ведению ИЛФ, принятое Американским торакальным обществом, Европейским респираторным обществом, Японским респираторным обществом и Латиноамериканской торакальной ассоциацией – An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management (2011) [1].
Радиологический паттерн ОИП
В новом руководстве представлена характеристика КТ-паттерна ОИП (таблица).

Наверное, не совсем правильно использовать термин «КТ-паттерн ОИП», поскольку термин «ОИП» относится к области патогистологии. По-видимому, более корректной является следующая формулировка: «При комбинации четырех признаков практически в 100% случаев наблюдается присутствие гистопатологического паттерна ОИП» [7].
Если все четыре признака присутствуют, хирургическая биопсия не проводится. Если имеется комбинация трех признаков, констатируется наличие возможной ОИП, требующей подтверждения путем биопсии. Наличие даже одного из семи перечисленных признаков несоответствия исключает диагноз ИЛФ.
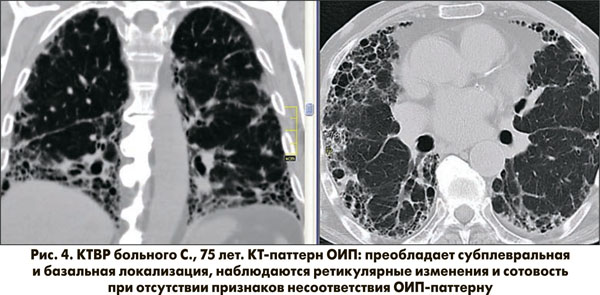
Рисунок 4 демонстрирует КТ-паттерн ОИП: на томограмме больного С. присутствуют все 4 признака.
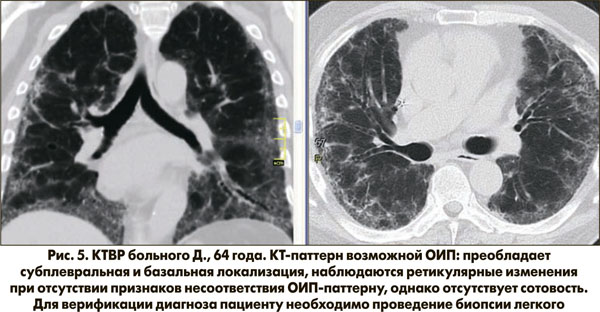
На рисунке 5 представлен КТ-паттерн возможной ОИП: на томограмме больного Д. присутствуют 3 признака – отсутствует сотовость, в связи с чем пациенту необходима верификация диагноза методом биопсии легкого.
Необходимо отметить, что «сотовое легкое» представляет конечную стадию многих интерстициальных болезней легких (end-stage lung) – гиперсенситивного пневмонита, саркоидоза легких, резистентного к глюкокортикостероидной и иммуносупрессивной терапии; других форм идиопатических интерстициальных пневмоний [8]. В связи с этим важное значение в диагностике ИЛФ имеет учет семи признаков несоответствия ОИП-паттерну, представленных в таблице. Следует отметить, что для исключения диагноза ИЛФ достаточно присутствия даже одного из этих признаков.
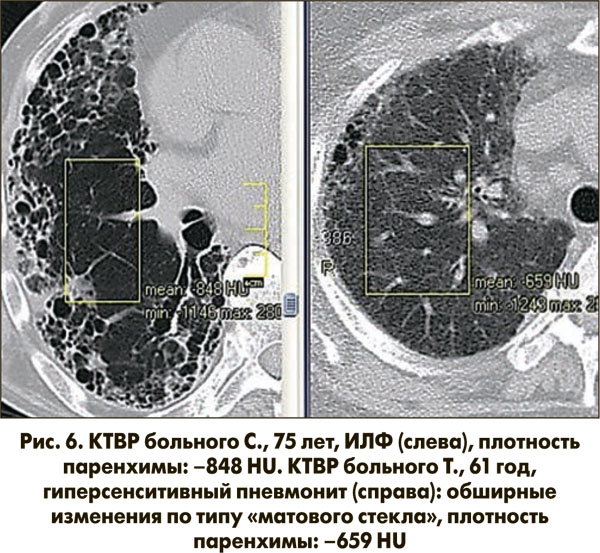
На рисунке 6 представлены КТВР двух пациентов с субплевральными сотовыми изменениями паренхимы легких. У пациента Т. (правая часть рисунка) определяется диффузное снижение прозрачности паренхимы (денситометрия: –659 HU) по типу «матового стекла», что исключает диагноз ИЛФ. На основании клинических и лабораторных данных больному был установлен диагноз гиперсенситивного пневмонита.
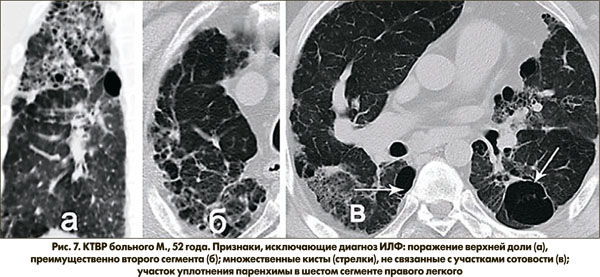
Рисунок 7 демонстрирует КТ-семиотику больного М., 52 лет, с клиническими признаками, совместимыми с диагнозом ИЛФ. Вместе с тем на КТВР определяются как минимум три признака, исключающие диагноз ИЛФ – поражение верхней доли (а), преимущественно второго сегмента (б); множественные кисты, не связанные с участками сотовости (в); участок уплотнения паренхимы в шестом сегменте правого легкого.
В значительной части случаев синдрома «сотового легкого» уже не удается установить его нозологическую принадлежность. Руководство по диагностике и ведению ИЛФ 2011 года [1] рекомендует в таких случаях в формуле диагноза использовать термин «неклассифицируемый фиброз легких» (МКБ‑10 J84.1).
Лабораторные и инструментальные методы
Результаты лабораторных исследований крови и жидкости бронхоальвеолярного лаважа не имеют диагностической ценности при ИЛФ.
При спирометрии и бодиплетизмографии определяются исключительно рестриктивные нарушения вентиляционной функции легких – уменьшение статических объемов и емкостей: общей емкости легких (TLC), остаточного объема (RV), жизненной емкости легких (VC), емкости вдоха (IC). Нарушения бронхиальной проходимости, как правило, несовместимы с диагнозом ИЛФ. Следует отметить, что скоростные показатели – форсированная жизненная емкость легких (FVC) и объем форсированного выдоха за первую секунду (FEV1) – обычно снижены, что объясняется падением TLC и VC. Однако индекс Генслера (FEV1/FVC) всегда увеличен, иногда до 90% и более, что характерно для рестриктивных нарушений.
Уже в ранних стадиях заболевания отмечается уменьшение диффузионной способности легких для монооксида углерода (DLCO). Необходимо подчеркнуть, что показатель DLCO является наиболее надежным в оценке темпов прогрессирования заболевания и определении эффективности проводимой терапии.
В заключение следует отметить, что в настоящее время наблюдается значительный рост интереса к проблеме ИЛФ со стороны крупных фармацевтических компаний. В последние годы синтезирован ряд принципиально новых антифибротических препаратов [9], увеличивается число многоцентровых рандомизированных исследований по их испытанию, в которых принимают участие и украинские научные центры. Все это создает основу для ожидания положительных результатов в решении проблемы своевременной диагностики и терапии ИЛФ – одной из наиболее трудных проблем современной пульмонологии.
Литература
1. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management // Am. J. Respir. Crit. Care Med. – 2011. – Vol. 183. – P. 788-824.
2. Fernandez-Perez E.R. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis / E.R. Fernandez-Perez, C.E. Daniels, D.R. Schroeder et al. // Chest. – 2010. – Vol. 137. – P. 129-137.
3. American Thoracic Society, European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement // Am. J. Respir. Crit. Care Med. – 2000. – Vol. 161. – P. 646-664.
4. Ідіопатичний легеневий фіброз: клініка, діагностика, лікування (проект національної угоди) / Ю.І. Фещенко, В.К. Гаврисюк, Є.О. Меренкова та ін. // Матер. V З’їзду фтизіатрів і пульмонологів України, 6-8 листопада 2013 р. – Київ, 2013. – С. 26-30.
5. Idiopathic Pulmonary Fibrosis / K.C. Meyer, S.D. Nathan, Editors. – Humana Press – brand of Springer, 2014. – 451 p.
6. Wuyts W.A. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? / W.A. Wuyts, A. Cavazza, G. Rossi et al. // Eur. Respir. Rev. – 2014. – Vol. 23. – P. 308-319.
7. Гаврисюк В.К. Эволюция принципов диагностики и терапии идиопатического легочного фиброза в положениях международных руководств // Очерки клинической пульмонологии. Под ред. В.К. Гаврисюка. – Киев, 2016. – С. 11-20.
8. American Thoracic Society/ European Respiratory Society. International Multidisciplinary Consensus on the Idiopathic Interstitial Pneumonias // Am. J. espir. Crit. Care Med. – 2002. – Vol. 165. – P. 277-304.
9. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline // Am. J. Respir. Crit. Care Med. – 2015. – Vol. 192. – P. e3-e19.