


16 листопада, 2016
Ингибирование PD-L1 в современной иммунотерапии злокачественных новообразований

Принципы, достижения, перспективы
Изучение иммуносупрессирующих свойств, проявляемых опухолями, а также поиск возможностей их коррекции привели к появлению нового класса таргетных препаратов. Сегодня проводится целый ряд исследований, в которых лиганд‑1 программируемой гибели клеток (PD-L1) выступает в качестве мишени для терапевтического воздействия при опухолях разных локализаций. PD-L1 также признан перспективным биомаркером для новообразований, которые характеризуются его избыточной экспрессией.
Иммунотерапия привлекает самое пристальное внимание исследователей во всем мире как перспективный подход к лечению опухолей, осуществляется поиск возможностей активировать естественные механизмы иммунной защиты для подавления опухолевого роста [1]. Во многом это связано с обнаружением следующего феномена: солидные опухоли и их микроокружение могут быть обильно инфильтрированы клетками иммунной системы, однако эти клетки не проявляют надлежащей активности. Возможность активировать эти клетки могла бы стать важным фактором в терапии онкологических заболеваний.
Обнаружение и уничтожение опухолевых клеток иммунной системой представляет собой сложный многоступенчатый процесс, в котором задействованы костимулирующие и корегуляторные молекулы (рис. 1).
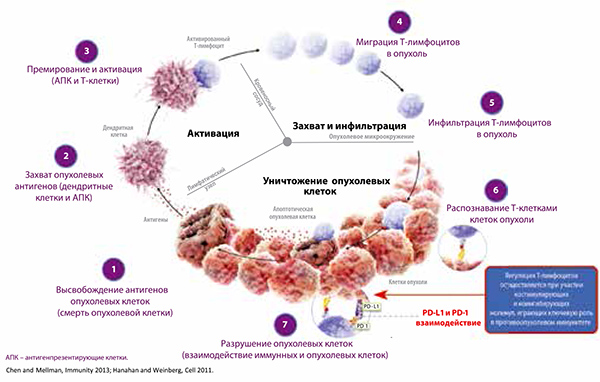 Рис. 1. Цикл иммунной защиты против опухоли
Рис. 1. Цикл иммунной защиты против опухолиКак известно, ведущую роль в обеспечении клеточного противоопухолевого иммунитета играют цитотоксические Т-лимфоциты, способные осуществлять лизис любых поврежденных клеток. Злокачественные клетки используют несколько механизмов ускользания от иммунного ответа, один из которых напрямую связан с гиперэкспрессией PD-L1 и подавлением активности Т-лимфоцитов (рис. 2).
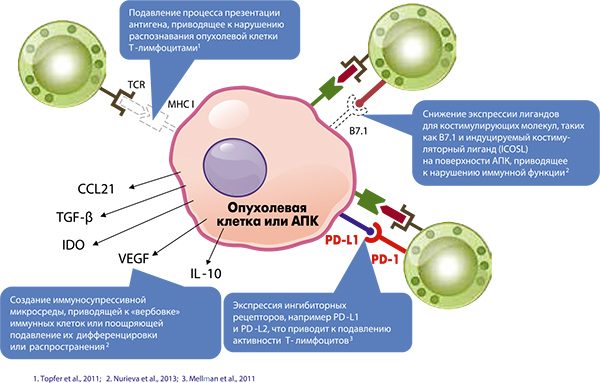 Рис. 2. Механизмы ускользания от иммунного надзора, используемые опухолевыми клетками
Рис. 2. Механизмы ускользания от иммунного надзора, используемые опухолевыми клеткамиПуть ускользания от иммунного надзора, опосредованный PD-L1/PD‑1
Сигнальный путь PD-L1/PD‑1 принимает участие в целом ряде сложных иммунных взаимодействий. Рецептор программируемой клеточной смерти PD1 представляет собой мембранный белок надсемейства иммуноглобулинов, который участвует в дифференцировке лимфоцитов и играет важную роль в отрицательной регуляции иммунной системы. Этот рецептор имеет два лиганда: PD-L1 и PD-L2. Помимо PD‑1, лиганд PD-L1 может также связываться с В7.1 – рецептором, присутствующим на поверхности активированных Т-лимфоцитов и дендритных клеток [2].
Иммунная толерантность регулируется несколькими отрицательными костимулирующими молекулами. Она связана с наличием PD1 и антигена CTLA‑4 на поверхности активированных Т-лимфоцитов, а также В7.1, B7.3 и лиганда PD-L1 на поверхности других клеток. В норме взаимодействие PD-L1/PD‑1 подавляет избыточную активацию иммунной системы и уменьшает Т-клеточную цитотоксичность, защищая здоровые клетки при хроническом воспалении. В организме человека PD-L1 экспрессируется на поверхности широкого диапазона клеток как иммунной системы (например, макрофагов), так и тканей организма (гематопоэтических, эпителиальных, эндотелиальных клеток), обеспечивая иммунную толерантность к ним [1].
Так называемая конститутивная иммунорезистентность опухоли связана с экспрессией PD-L1 на поверхности злокачественных клеток. Этот лиганд взаимодействует с PD‑1, присутствующим на поверхности активированных клеток иммунной системы (Т- и В-лимфоцитов, натуральных киллеров, моноцитов, дендритных клеток). Активация PD‑1 путем связывания с PD-L1 приводит к ингибированию сигнального пути цитотоксических Т-лимфоцитов, нарушает в них процессы транскрипции и подавляет механизмы иммунной защиты [3, 4]. Параллельно подавляются процессы в иммунной системе, связанные с активностью других видов иммунных клеток, экспрессирующих PD‑1.
Кроме того, сигналом для экспрессии PD-L1 опухолевыми клетками может послужить повышенная продукция интерферона (ИФН-g) при воспалении, сопутствующем росту новообразования. То есть экспрессия PD-L1 может быть индуцирована со временем адаптивным ответом на воспалительный процесс (адаптивная иммунорезистентность) [3].
Для создания иммунодепрессивной микросреды, благоприятной для опухолевого роста, злокачественные новообразования секретируют множество регуляторных молекул. Опухоли продуцируют макрофагальный колониестимулирующий фактор, цитокины IL‑6, IL‑10, ганглиозиды и другие факторы, подавляющие развитие дендритных клеток; тот же IL‑10, а также индоламин‑2,3-диоксигеназа (IDO) способны ингибировать процесс активации Т-лимфоцитов. Выделяемые опухолью IL-1β, фактор роста эндотелия сосудов (VEGF) и колониестимулирующий фактор гранулоцитов и макрофагов (GM‑CSF) могут вызвать пролиферацию иммуносупрессивных миелоидных клеток [5].
Сосудистые изменения в микроокружении, связанные с высвобождением VEGF, приводят к уменьшению инфильтрации опухоли T-лимфоцитами. Патологическая секреция трансформирующего фактора роста (TGF-β) способствует превращению наивных CD4+ Т-клеток в регуляторные Т-лимфоциты и подавлению Т-клеточной активности в ложе опухоли (как и активности АПК). Активация Т-клеток при отсутствии надлежащей костимуляции приводит к анергии – потере Т-лимфоцитами способности отвечать на антигенную стимуляцию [5].
Целью иммунотерапии является повышение общего иммунного ответа на опухоль и преодоление иммунной толерантности раковых клеток. С этой целью предпринимаются попытки воздействовать на разные этапы цикла активации Т-лимфоцитов, используя антитела против PD-L1, PD‑1, CTLA‑4, ингибиторы VEGF, IDO, GM-CSF, противоопухолевые вакцины и т.д.
Роль ингибиторов PD-L1 в клинической практике
О наличии опухоль-инфильтрирующих иммунных клеток (рис. 3) сообщалось при многих типах злокачественных новообразований с гиперэкспрессией PD-L1 [1, 6-9]. В целом ряде научных работ указывалось, что избыточная экспрессия PD-L1 может способствовать усилению опухолевого роста и метастазирования [1, 3, 10]. Пациенты с опухолями, экспрессирующими PD-L1, вероятно, имеют неблагоприятный прогноз. Кроме того, экспрессия PD-L1 в опухолевых клетках связана с более поздней стадией заболевания или степенью злокачественности опухоли, является важным прогностическим и предиктивным фактором для некоторых видов опухолей [10].
Избыточная экспрессия PD-L1 выявляется в клетках более чем 50% опухолей человека [1, 6-9], причем наиболее характерна для следующих новообразований: глиобластома и смешанная глиома (встречается в 100% случаев), назофарингеальная карцинома (68-100%), меланома (40-100%), множественная миелома (93%), лимфома (17‑94%), опухоли мочевого пузыря (28-100%). Кроме того, она выявляется при немелкоклеточном раке легкого (35-95%; рис. 3), аденокарциномах кишечника (53%), гепатоцеллюлярной карциноме (45-93%), раке яичника (33-80%), раке поджелудочной железы (39%), злокачественных опухолях желудка и пищевода (42%). Несколько реже встречаются случаи гиперэкспрессии PD-L1 при почечноклеточной карциноме (15‑24%), раке грудной железы (31-34%), а также при лейкемиях (11-42%) [1, 6]. Важно, что при всех этих заболеваниях (включая Т-клеточные лимфомы, диффузную В-крупноклеточную лимфому и лимфому из малых лимфоцитов) опухоли инфильтрированы иммунными клетками.
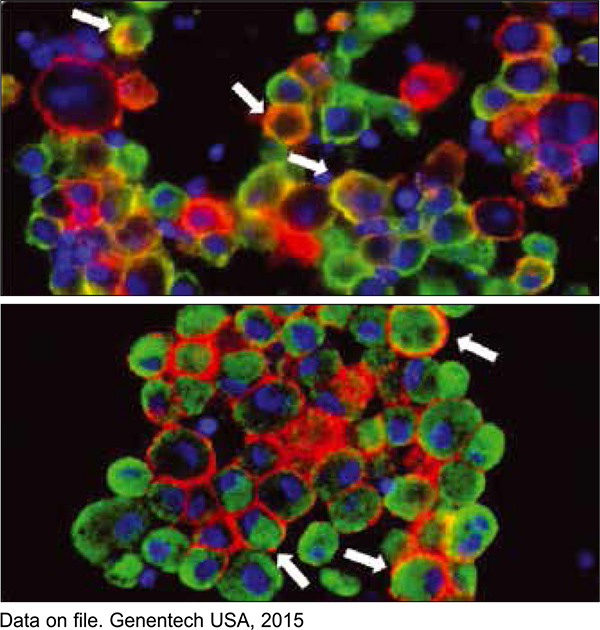 Рис. 3. Экспрессия PD-L1, выявленная иммунофлюоресцентным методом на клетках рака легкого (красный цвет). Стрелками показаны инфильтрирующие опухоль макрофаги
Рис. 3. Экспрессия PD-L1, выявленная иммунофлюоресцентным методом на клетках рака легкого (красный цвет). Стрелками показаны инфильтрирующие опухоль макрофаги(зеленые: CD163 на верхнем снимке, CD68 на нижнем), которые экспрессируют PD-L1.
Ядра клеток обозначены синим
В целом ряде исследований показано, что антитела против PD-L1 способны активировать цитотоксические Т-лимфоциты, делая опухоль более уязвимой для иммунной системы [6, 11-18]. Важно, что антитела против PD-L1 предотвращают подавление Т-клеточной активности, опосредованное PD‑1 и В7.1; при этом обеспечивается сохранность взаимодействия PD-L2/PD‑1, что способствует профилактике аутоиммунных процессов. Подавление PD‑1 оказалось менее эффективным и сопряжено с повышенным количеством побочных эффектов, поскольку приводит к блокированию взаимодействия с обоими лигандами (PD-L1 и PD-L2) этого рецептора, увеличивая вероятность серьезных аутоиммунных реакций [6]. Напротив, сохранение взаимодействия PD‑1 с PD-L2 позволяет поддерживать иммунный гомеостаз в здоровых тканях организма.
Таким образом, целенаправленное подавление активности PD‑1/PD-L1 на опухолевых клетках, а также иммунных клетках, инфильтрирующих опухоль, предотвращает сигнальное взаимодействие, опосредованное В7.1 и PD‑1 (рис. 4) [19]. Предотвращение связывания PD-L1 с его рецепторами на Т-лимфоцитах делает возможным возвращение активности последних: еще доклинические исследования убедительно показали, что предотвращение двух упомянутых видов взаимодействия увеличивает Т-клеточную цитотоксичность.
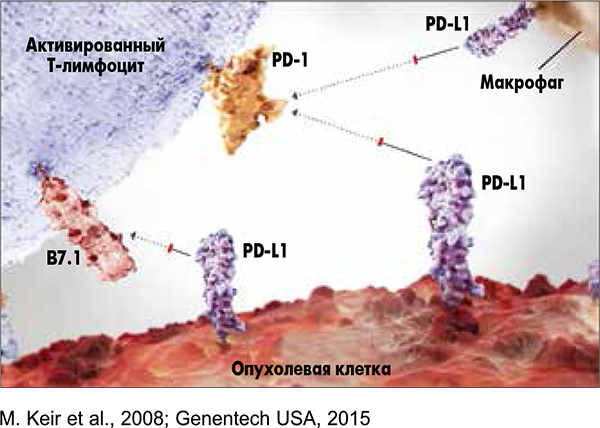 Рис. 4. PD-L1 на поверхности клеток. Варианты взаимодействий с рецепторами
Рис. 4. PD-L1 на поверхности клеток. Варианты взаимодействий с рецепторамиВ настоящее время на стадии разработки находится целый ряд препаратов на основе антител против PD-L1 и PD‑1, отдельные из которых уже разрешены к применению в клинической практике.
Ингибитор PD-L1 атезолизумаб (MPDL3280A) представляет собой высокотехнологический таргетный препарат, разработанный компанией Genentech. Это генно-инженерное антитело (IgG1) против PD-L1, специально модифицированное путем замены аминокислоты (N298A) в Fc-фрагменте тяжелых цепей молекулы иммуноглобулина, чтобы исключить инициирование антителозависимой клеточной цитотоксичности в отношении Т-лимфоцитов и других нормальных клеток организма, экспрессирующих PD-L1 [11].
Доклинические исследования на различных экспериментальных моделях показали эффективность антитела MPDL3280A как в отношении селективного подавления PD-L1/PD‑1 взаимодействий, так и активации цитотоксической Т-клеточной активности. Кроме того, в экспериментах с использованием сингенных опухолевых моделей показаны различия в эффективности антител MPDL3280A при их применении с разными группами химиопрепаратов (рис. 5). Уничтожение опухолевых клеток химиотерапевтическим препаратом может привести иммунную систему к более высоким уровням опухолевых антигенов и потенциально увеличить презентацию опухолевых антигенов (Hodge et al., 2013; Chen and Mellman, 2013).
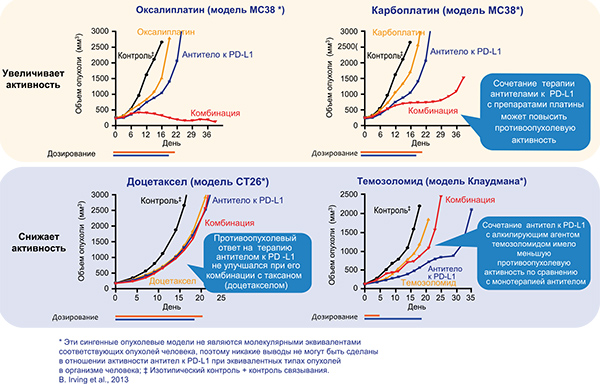 Рис. 5. Различия в эффективности комбинированного применения антител против PD-L1 с химиопрепаратами
Рис. 5. Различия в эффективности комбинированного применения антител против PD-L1 с химиопрепаратамиПомимо цитотоксической активности, некоторые химиотерапевтические препараты обладают иммуногенным эффектом (Zitvogel et al., 2008). Так, оксалиплатин может индуцировать иммуногенную клеточную смерть (Tesniere et al., 2010; Michaud et al., 2011; Green et al., 2009), а 5-фторурацил может вызвать селективное восстановление миелоидных супрессорных клеток и увеличить производство IFN-γ (Vincent et al., 2010). Поддерживая первый этап противоопухолевого иммунитета (высвобождение антигенов опухолевых клеток при их разрушении), химиотерапия может улучшить иммунный ответ за счет повышения дальнейшей блокады PD-L1/PD-1. В исследовании фазы Ib (GP28328) изучается эффективность атезолизумаба в сочетании с препаратом платины при немелкоклеточном раке легкого.
Атезолизумаб стал первым препаратом в классе ингибиторов PD-L1, который одобрен для применения в терапии пациентов с распространенным раком мочевого пузыря. Изучению этого препарата посвящено базовое исследование II фазы IMvigor 210, предварительные результаты которого были озвучены в 2015 г. (результаты исследования отображены в пресс-релизе «FDA одобрило применение атезолизумаба у пациентов с распространенной формой уротелиального рака мочевого пузыря, ранее не получавших лечения». – Прим. ред.). В настоящее время продолжается рандомизированное исследование III фазы IMvigor 211, в котором атезолизумаб сравнивается со стандартной химиотерапией у пациентов с метастатической уротелиальной карциномой, прогрессирующей после первоначального лечения. Все исследования включают в себя оценку сопутствующего теста, разработанного для определения статуса PD-L1.
Кроме того, в настоящее время атезолизумаб по ускоренной процедуре рассматривается Управлением по контролю качества пищевых продуктов и лекарственных препаратов США (FDA) для лечения пациентов с PD-L1 положительным немелкоклеточным раком легкого (результаты исследования отображены в пресс-релизе «Атезолизумаб (Тецентрик®, Roche) показал увеличение продолжительности жизни пациентов с определенным типом рака легкого в сравнении с пациентами, получавшими химиотерапию, согласно результатам исследования III фазы». – Прим. ред.).
В целом открытие возможности селективного подавления PD-L1 дало начало новому классу таргетных препаратов. Ввиду особенностей терапевтического воздействия ингибиторов PD-L1 (стимуляция противоопухолевого иммунного ответа) и большого разнообразия новообразований, экспрессирующих PD-L1, этот новый класс препаратов может принципиально изменить облик современной клинической онкологии.
Перспективы совместного применения с другими таргетными препаратами
Как известно, современные таргетные подходы направлены на ингибирование конкретных сигнальных путей или мутантных белков, которые имеют решающее значение для роста или выживания опухоли. Несмотря на высокую эффективность таргетных препаратов, в большинстве случаев к ним со временем развивается резистентность. Добавление антител против PD-L1 к терапии ингибиторами сигнальных путей МАРК, МЕК, BRAF и т.д. способно усиливать противоопухолевые иммунные реакции, а также стать эффективным подходом для профилактики или преодоления резистентности.
Известно, что активация MEK увеличивает экспрессию PD-L1 опухолевыми клетками, усиливая иммунную толерантность и устойчивость к MEK-ингибиторам [12]. Ингибирование MEK может увеличивать экспрессию молекул главного комплекса гистосовместимости (MHC) I класса на поверхности клеток опухоли, облегчая их ускользание от иммунного надзора [13].
Ингибирование BRAF повышает экспрессию антигенов, ассоциированных с меланомой, а также уменьшает секрецию иммуносупрессивных цитокинов [14] и может усиливать Т-клеточную инфильтрацию ложа опухоли [15]. В исследованиях I фазы изучалась эффективность атезолизумаба в сочетании с вемурафенибом при меланоме с мутацией BRAF V600, а также в сочетании с кобиметинибом при KRAS-мутантных опухолях кишечника, немелкоклеточном раке легкого, меланоме и других солидных опухолях. Получены обнадеживающие результаты, в частности усиление Т-клеточной активности и увеличение количества цитотоксических Т-лимфоцитов. В итоге МЕК-ингибиторы продемонстрировали явный синергетический эффект при применении с ингибиторами PD-L1 на некоторых моделях опухолевых клеток [13].
Как известно, увеличение смертности в популяции опухолевых клеток связано с более эффективным высвобождением и презентацией опухолевых антигенов. Указывается, что ингибиторы EGFR могут влиять на иммунные или воспалительные реакции путем прямой модуляции экспрессии MHC и потенциально способны активировать эффекторный Т-клеточный ответ. Как показали B.P. Pollack и соавт. (2011), сочетание атезолизумаба с эрлотинибом (препаратом, приводящим к высвобождению антигенов и позитивной модуляции MHC I) может обеспечивать дополнительную и продолжительную противоопухолевую эффективность [16].
Избыточная секреция VEGF способствует не только опухолевому ангиогенезу, но и развитию иммунной толерантности посредством нескольких различных механизмов, в частности, уменьшает адгезию лимфоцитов к стенкам сосудов и инфильтрацию ими опухоли [17], ухудшает функцию дендритных клеток [18].
Ингибирование VEGF нормализует состояние локальной сосудистой сети, усиливает активность Т-лимфоцитов и инфильтрации ложа опухоли [17, 20]. В связи с этим комбинированное применение антител против PD-L1 и анти-VEGF препарата (бевацизумаба) может усилить противоопухолевый иммунный ответ, приводя к более стойкому клиническому эффекту. В исследованиях II фазы изучается эффективность применения сочетания атезолизумаба с бевацизумабом и режимом химиотерапии FOLFOX при солидных опухолях; комбинации атезолизумаба с бевацизумабом в сравнении с монотерапией атезолизумабом или монотерапией сунитинибом при почечноклеточном раке.
Ингибирование PD-L1 предотвращает инактивацию цитотоксических Т-лимфоцитов, способствует восстановлению их активности и усиливает реакцию иммунной системы на опухоль. При этом в здоровых тканях организма сохраняется иммунный гомеостаз (за счет сохранного взаимодействия PD‑1 с PD-L2).
В настоящее время ингибиторы PD-L1, в частности атезолизумаб, интенсивно изучаются как в монорежимах, так и в сочетаниях с таргетными препаратами других групп и химиотерапией при опухолях разных локализаций и молекулярных подтипов. Атезолизумаб разрешен к применению в США при раке мочевого пузыря и проходит рассмотрение по ускоренной процедуре FDA для применения при раке легкого.
Литература
1. Chen D., Mellman I. Immunity. 2013; 39: 1-10.
2. Mellman I., Coukos G., Dranoff G. Nature. 2011; 480: 480-9.
3. Merelli B. et al. Crit Rev Oncol Hematol. 2014; 89: 140-65.
4. Pardoll D.M. Nature Rev Cancer. 2012; 12: 252-64.
5. Andersen M.H. Cancer Immunol Immunother. 2012; 61: 1289-97.
6. Chen D., Irving B., Hodi F. Clin Cancer Res. 2012; 18: 6580-6587.
7. Hanahan D., Weinberg R. Cell. 2011.
8. Nelson D. et al. J. Immunol. Res. 2014.
9. Quesada S., Peggs K. Br. J. Cancer. 2013.
10. Choueiri T.K. et al. Annals of Oncology 25(11): 2178-84, 2014.
11. Herbst R.S. et al. ASCO. 2013.
12. Yamamoto R. et al. Cancer Sci. 2009; 100: 2093-100.
13. Irving B.A. et al. Soc Immunother Cancer. 2013.
14. Vannemann M., Dranoff G. Nature Rev Cancer. 2012; 12: 237-50.
15. Liu C. et al. Clin Cancer Res. 2013; 19: 393-403.
16. Pollack B.P. et al. Clin Cancer Res. 2011; 17: 4400-13.
17. Bouzin C. et al. J Immunol. 2007; 178: 1505-11.
18. Huang Y. et al. Blood. 2007; 110: 624-31.
19. Keir M. et al. Annu. Rev. Immunol. 2008.
20. Shrimali R.K. et al. Cancer Res. 2010; 70: 6171-80.
Подготовила Катерина Котенко