


25 листопада, 2015
Хронический лимфолейкоз / лимфома из малых лимфоцитов
Хронический лимфолейкоз (ХЛЛ)/ лимфома из малых лимфоцитов (ЛМЛ) – опухолевое заболевание системы крови, характеризующееся пролиферацией и накоплением в крови, костном мозге и лимфоидных органах морфологически зрелых и иммунологически некомпетентных В-лимфоцитов, имеющих характерный иммунофенотип. ХЛЛ и ЛМЛ являются различными манифестациями одного и того же заболевания. В обоих случаях основным субстратом являются клональные малые В-лимфоциты. Отличия заключаются в том, что основная масса опухолевых лимфоцитов при ХЛЛ сосредоточена в костном мозге и периферической крови, а при ЛМЛ – в лимфатических узлах.
Диагностика
Диагноз ХЛЛ требует наличия ≥5000 клональных В-клеток/мкл (5×109/л) в периферической крови, установленного по данным флоуцитометрии. Наличие меньшего количества В-клеток при отсутствии пальпируемых лимфоузлов и других клинических признаков лимфопролиферативного заболевания обозначается как моноклональный В-лимфоцитоз (МВЛ). У пациентов с МВЛ часто наблюдаются благоприятные в молекулярном плане очаги, мутированный ген IGHV, хромосомная аномалия del(13q) или нормальная цитогенетика. Вероятность прогрессирования МВЛ в ХЛЛ составляет 1,1% в год. Диагноз ЛМЛ устанавливается при наличии лимфаденопатии и/или спленомегалии с содержанием В-лимфоцитов в периферической крови <5×109/л.
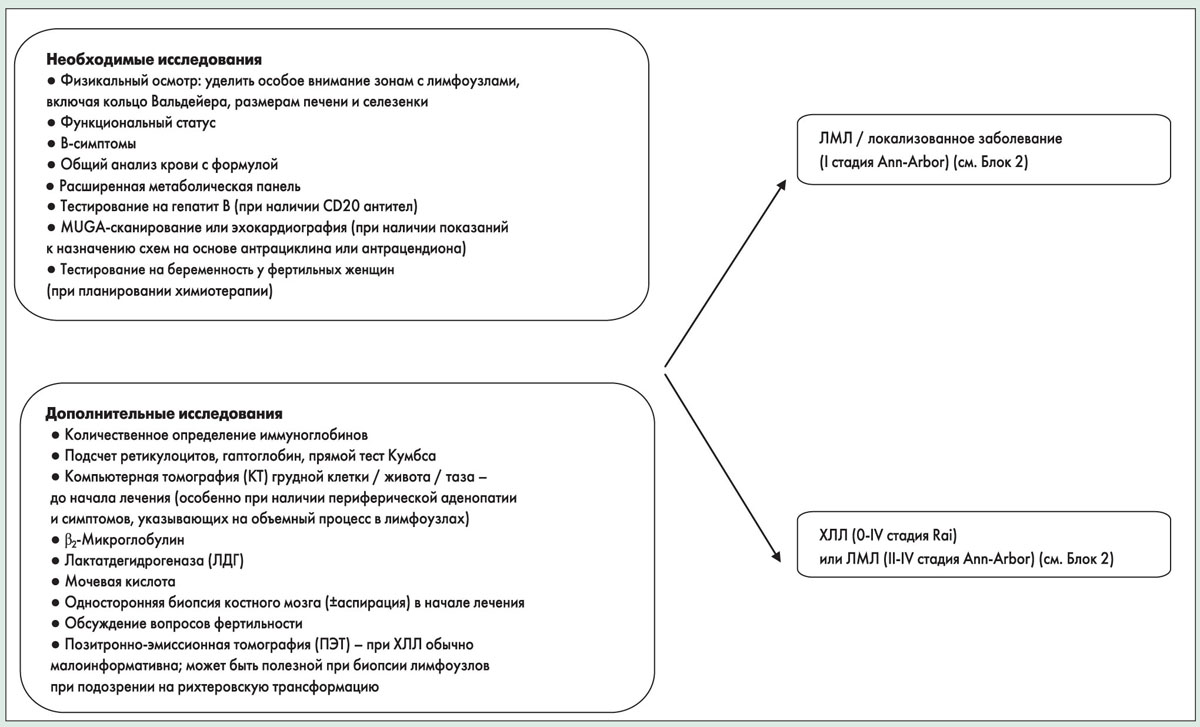
Блок 1. Первичная диагностика
Необходимым этапом диагностики ХЛЛ/ЛМЛ является иммунофенотипирование. В случае ХЛЛ, как правило, достаточно флоуцитометрии периферической крови, биопсия костного мозга обычно не требуется. Диагноз ЛМЛ в идеале должен быть подтвержден с помощью биопсии лимфоузла. Поверхностные клеточные маркеры для флоуцитометрических исследований должны включать каппа/лямбда, CD19, CD20, CD5, CD23 и CD10. Если флоуцитометрия используется для установления диагноза, дополнительно следует оценить циклин D1 или наличие t(11;14) для исключения лимфомы из клеток мантии. Если флоуцитометрия не позволила установить диагноз, проводят иммуногистохимическое исследование парафиновых срезов. Рекомендованная иммуногистохимическая панель включает CD3, CD5, CD10, CD20, СD23 и циклин D1.
По последним данным, комплексный кариотип (≥3 не связанных хромосомных аномалий более чем в 1 клетке при обычном кариотипировании стимулированных клеток ХЛЛ) ассоциируется с неблагоприятным прогнозом.
Прогностические факторы
За последнее десятилетие был идентифицирован ряд факторов, имеющих прогностическую значимость у пациентов с ХЛЛ, в том числе сывороточные (тимидинкиназа, β2-микроглобулин) и генетические маркеры (мутационный статус IGHV).
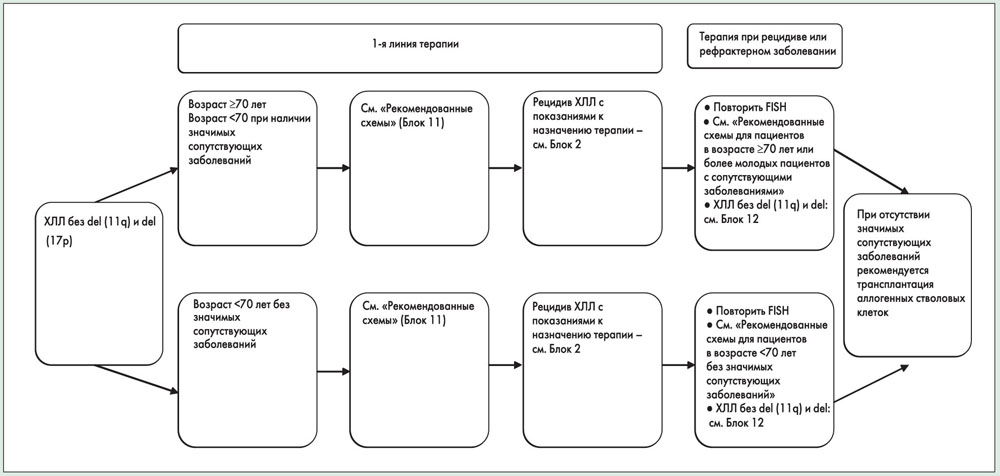
Блок 4. ХЛЛ без делеций 11q и 17p
Мутационный статус гена IGHV (вариабельного региона тяжелой цепи иммуноглобулина) является важным предиктором выживаемости. Немутированный IGHV (≥98% гомологичность с геном зародышевой линии) ассоциируется с неблагоприятным прогнозом и значительно сниженной выживаемостью по сравнению с мутированным IGHV независимо от стадии заболевания. Кроме того, вовлечение гена VH3-21 ассоциируется с неблагоприятными исходами независимо от мутационного статуса.
Экспрессия CD38 (≥7% В-лимфоцитов) и/или ZAP70 (≥20% В-лимфоцитов) связана с более низкими показателями выживаемости без прогрессирования (ВБП) и общей выживаемости (ОВ).
Среди флоуцитометрических маркеров CD49d является наиболее сильным прогностическим параметром и единственным маркером, не зависящим от результатов FISH и статуса IGHV.
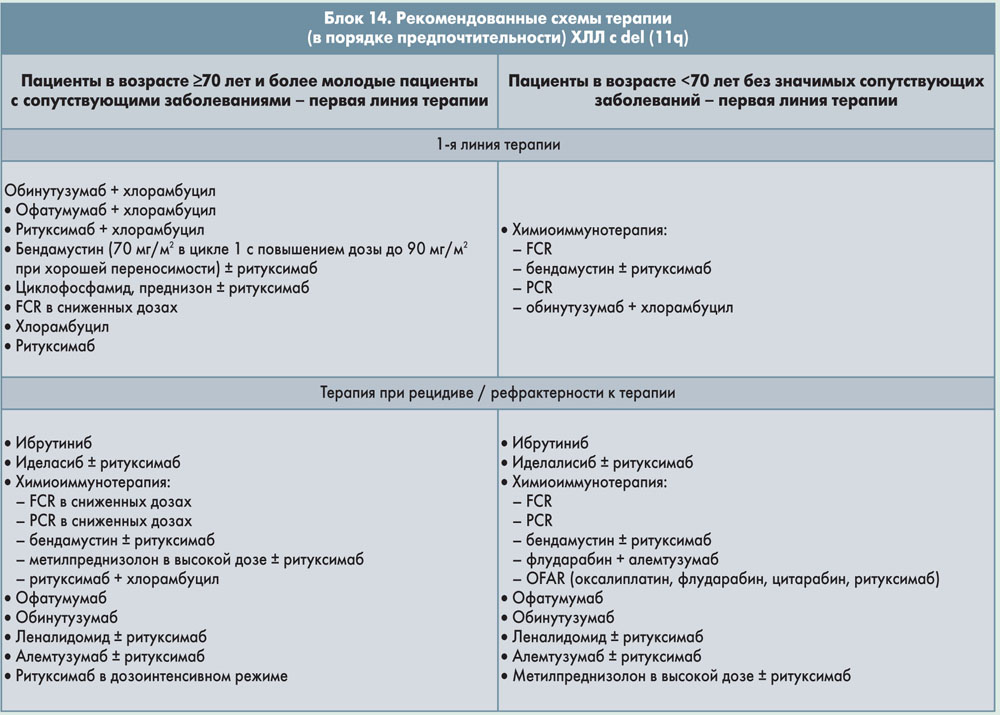
Блок 6. ХЛЛ с делецией 11q
CD38 и ZAP70 положительно коррелируют с немутированным IGHV и могут использоваться как суррогатные маркеры мутационного статуса IGHV.
Повышенные уровни β2-микроглобулина являются сильным независимым предиктором интервала без лечения, ответа на терапию и ОВ, в том числе у пациентов, получающих 1-ю линию химиоиммунотерапии. Важным преимуществом β2-микроглобулина является простота определения, однако следует учитывать влияние почечной дисфункции.
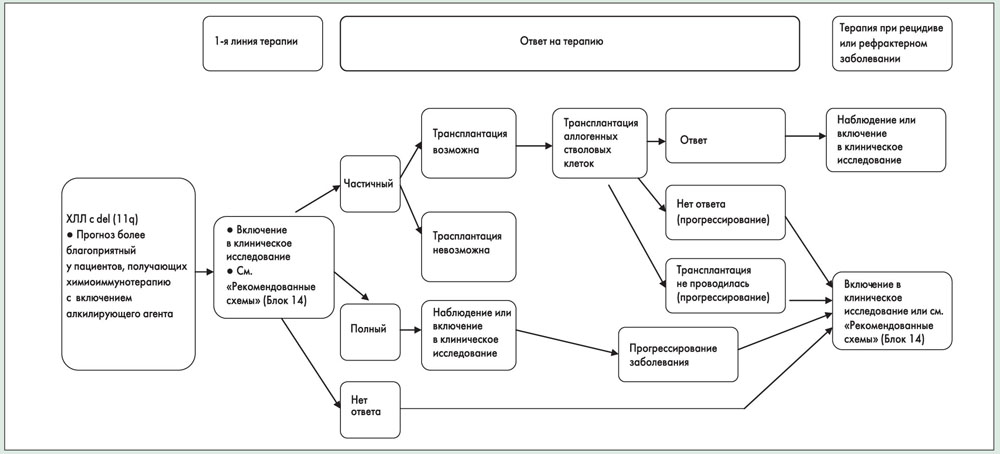
 Цитогенетические аномалии, выявляемые с помощью FISH, присутствуют более чем у 80% пациентов с ХЛЛ, не получавших лечения. Наиболее частые аномалии – del(13q) (55%) как единственное отклонение, затем следуют del(11q) (18%), трисомия 12 (16%), del(17p) (7%) и del(6q) (7%). Del(13q) как единственная аномалия ассоциируется с благоприятным прогнозом и наиболее длительной медианой выживаемости (133 мес). Del(11q) часто ассоциируется с выраженной лимфаденопатией, прогрессированием заболевания и более короткой медианой выживаемости (79 мес). У пациентов с del(11q) и полной утратой функции гена ATM может наблюдаться сниженный ответ на лучевую терапию и цитотоксические препараты, что отражается неблагоприятным клиническим исходом.
Цитогенетические аномалии, выявляемые с помощью FISH, присутствуют более чем у 80% пациентов с ХЛЛ, не получавших лечения. Наиболее частые аномалии – del(13q) (55%) как единственное отклонение, затем следуют del(11q) (18%), трисомия 12 (16%), del(17p) (7%) и del(6q) (7%). Del(13q) как единственная аномалия ассоциируется с благоприятным прогнозом и наиболее длительной медианой выживаемости (133 мес). Del(11q) часто ассоциируется с выраженной лимфаденопатией, прогрессированием заболевания и более короткой медианой выживаемости (79 мес). У пациентов с del(11q) и полной утратой функции гена ATM может наблюдаться сниженный ответ на лучевую терапию и цитотоксические препараты, что отражается неблагоприятным клиническим исходом.
Недавно было установлено, что ранее не леченные пациенты с del(11q) хорошо отвечают на комбинированную терапию флударабином и циклофосфамидом. Следовательно, добавление алкилирующего агента к флударабину может ослаблять неблагоприятную прогностическую значимость del(11q).
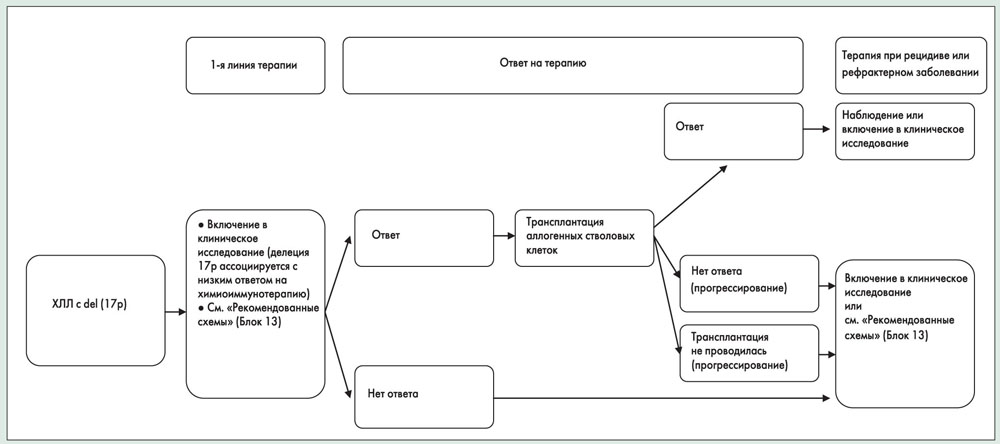
Аномалия del(17p), которая отражает утрату гена ТР53 и часто сопровождается мутациями оставшейся аллели ТР53, ассоциируется с неблагоприятными исходами – коротким интервалом без лечения, короткой медианой выживаемости (32 мес) и низким ответом на химиотерапию.
Мутации гена ТР53 могут наблюдаться при отсутствии del(17p) и являются независимым предиктором сниженной выживаемости и повышенной резистентности к химиотерапии.
Мутации генов NOTCH1, SF3B1 и BIRC3 обнаруживаются у 4-15% пациентов с впервые диагностированной ХЛЛ. У больных, рефрактерных к флударабину, они наблюдаются намного чаще (в 15-25% случаев). Интегрированная прогностическая модель, включающая эти новые маркеры, позволяет классифицировать пациентов на 4 группы: высокий риск (аномалии ТР53 и/или BIRC3), промежуточный риск (мутации NOTCH1 и/или SF3b1, и/или наличие del(11q), низкий риск (наличие только трисомии 12) и очень низкий риск (наличие только del(13q). Средняя 10-летняя выживаемость для этих групп составляет 29, 37, 57 и 69% соответственно.

Мутация NOTCH1 независимо ассоциируется с рихтеровской трансформацией (45 vs 5% без мутации через 15 лет; р<0,001).
Обследование
Диагностические процедуры при ХЛЛ/ЛМЛ не отличаются от таковых при других лимфоидных опухолях. Количественное определение иммуноглобулинов может быть информативным у пациентов с частыми инфекциями. Характер вовлечения костного мозга (диффузный vs очаговый) имеет прогностическую значимость, однако в настоящее время не оценивается, учитывая наличие более значимых иммуногистохимических и флоуцитометрических маркеров, которые можно оценить с помощью анализа периферических лимфоцитов.
Компьютерная томография (КТ) помогает мониторировать прогрессирование заболевания у пациентов с новыми симптомами, у которых периферическая лимфаденопатия отсутствует. У бессимптомных больных периодическое КТ-исследование не рекомендуется.
У пациентов с анемией необходимы подсчет ретикулоцитов и прямой тест Кумбса для исключения возможности гемолиза и «чистой» формы аплазии системы красной крови.
ПЭТ-сканирование при ХЛЛ малоинформативно, но может помочь выбрать лимфоузел для биопсии при подозрении на рихтеровскую трансформацию.

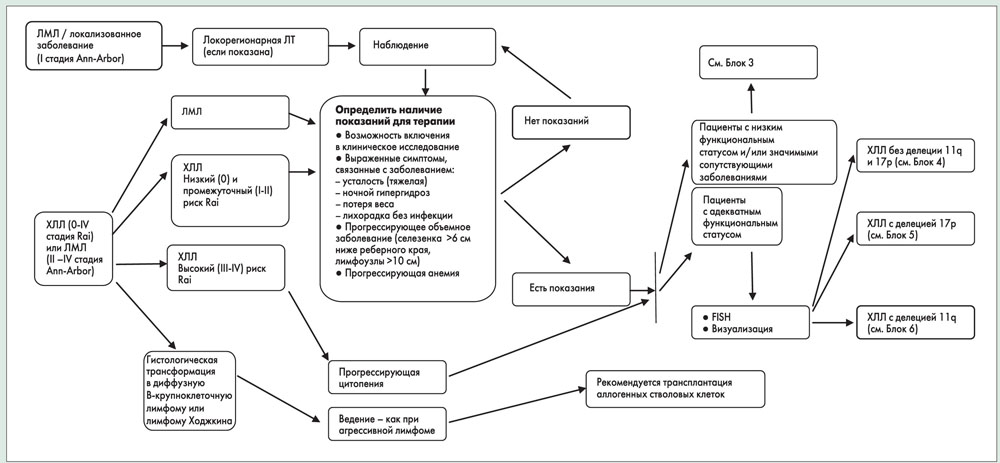
Определение стадии заболевания
В настоящее время во всем мире для определения стадии заболевания у пациентов с ХЛЛ используются две системы – Rai и Binet. Обе системы основаны исключительно на данных физикального обследования (наличие вовлечения лимфоузлов, гепато- и/или спленомегалии) и параметрах крови (наличие анемии или тромбоцитопении). Выживаемость пациентов с заболеванием низкого риска (Rai 0; медиана выживаемости 150 мес) практически не отличается от таковой в общей популяции аналогичного возраста. Медиана выживаемости пациентов с заболеванием промежуточного риска (Rai I-II) составляет 71-101 мес, с заболеванием высокого риска (Rai III-IV) – 19 мес.
Критерии ответа
Критерии ответа на лечение представлены в блоке 10. Для полного ответа должны быть соблюдены все критерии (оцениваются не ранее чем через 2 мес после завершения лечения): содержание лимфоцитов в периферической крови <4×109/л, отсутствие лимфаденопатии (пальпируемые лимфоузлы ≤1,5 см в диаметре); отсутствие мегалии; отсутствие спленомегалии; отсутствие конститутивных симптомов (снижения веса, выраженной патологической усталости, лихорадки, повышенной ночной потливости) и нормализация показателей крови без введения факторов роста (нейтрофилы >1,5×109/л, тромбоциты >100×109/л, гемоглобин >11 г/дл). При частичном ответе необходимо наличие по крайней мере 2 критериев из следующих: снижение на ≥50% по сравнению с исходным содержания лимфоцитов в периферической крови, лимфаденопатии, гепатомегалии и/или спленомегалии; кроме того, по крайней мере 1 показатель крови должен нормализоваться или улучшиться на ≥50% по сравнению с исходным, и это улучшение должно сохраняться не менее 2 мес.
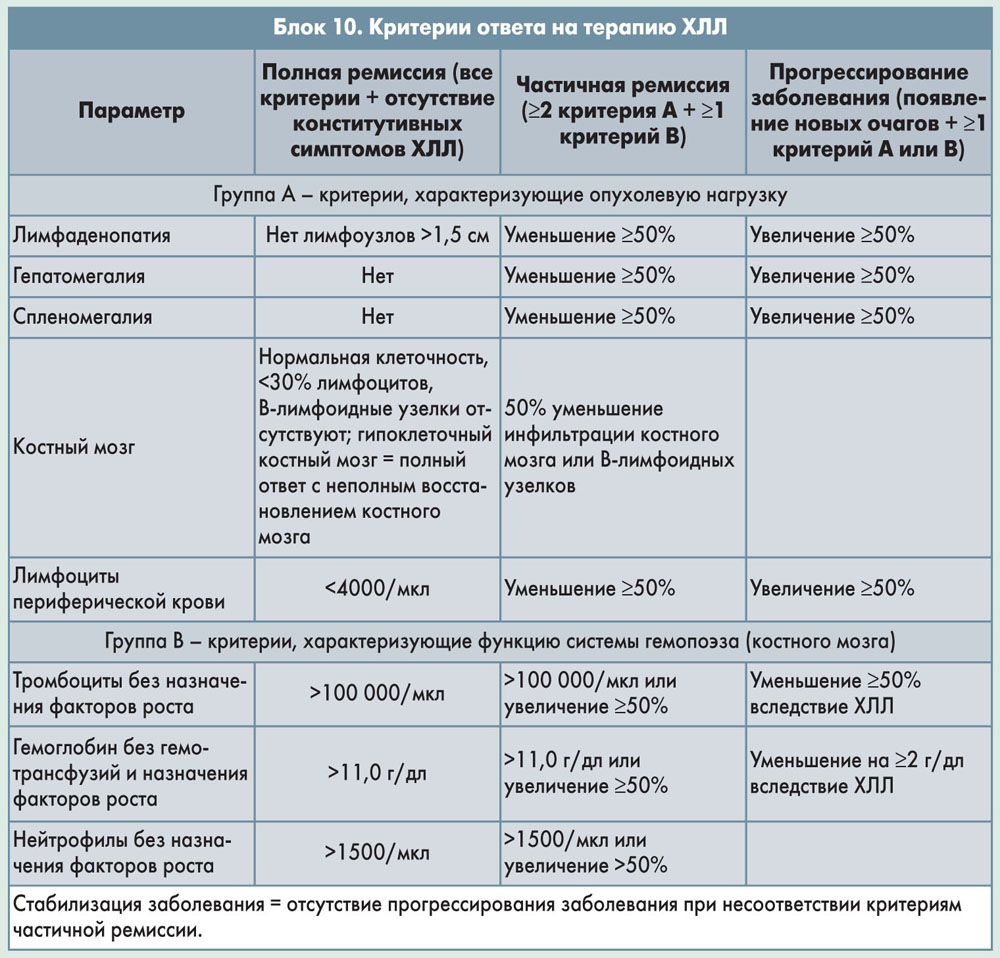
Прогрессирование заболевания определяется при наличии любого признака из следующих: повышение на ≥50% по сравнению с исходным содержания лимфоцитов в периферической крови, лимфаденопатии, гепатомегалии и/или спленомегалии; появление новых очагов; появление цитопении, связанной с заболеванием (снижение тромбоцитов на ≥50%, снижение гемоглобина на >2 г/дл по сравнению с исходным). Стабилизация заболевания определяется как отсутствие прогрессирования заболевания при несоответствии критериям полного и частичного ответа.
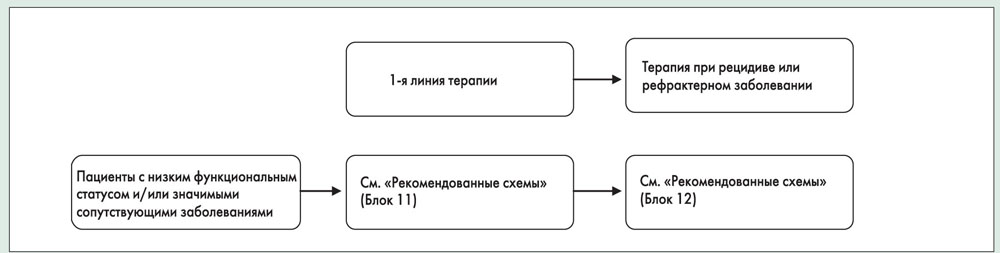
Лечение
Схемы лечения в зависимости от стадии заболевания и функционального статуса пациента представлены в блоках 11-14.

Гистологическая трансформация
У 2-10% пациентов с ХЛЛ при естественном течении заболевания или во время лечения развивается рихтеровская трансформация (гистологическая трансформация в диффузную В-крупноклеточную лимфому или лимфому Ходжкина). Частота гистологической трансформации тем выше, чем больше схем терапии использовалось. Вероятными генетическими путями, вовлеченными в патогенез рихтеровской трансформации, являются инактивация NOTCH1 и нарушения TP53 и CDKN2A/B.

Пациенты с рихтеровской трансформацией должны получать химиоиммунотерапию по схемам, изначально разработанным для диффузной В-крупноклеточной лимфомы. Кроме того, могут применяться схемы OFAR и гипер-CVAD с ритуксимабом. Пациентам, ответившим на первичное лечение, рекомендуется трансплантация аллогенных стволовых клеток.

Пациенты с ходжкинской гистологией должны получать стандартные схемы, применяющиеся при лимфоме Ходжкина.
Другие гистологические трансформации, включая ХЛЛ с повышенными пролимфоцитами и ускоренную ХЛЛ (наличие центров расширенной пролиферации или высокая скорость пролиферации), ассоциируются с более агрессивным течением заболевания; оптимальное ведение не разработано.
Руководство печатается в сокращении.
Полная версия на www.nccn.org
Перевел с англ. Алексей Терещенко