


5 листопада, 2016
Лабораторная диагностика заболеваний поджелудочной железы
Основными прогностическими тестами (маркерами панкреонекроза) при ОП являются СРБ (чувствительность – 86%), эластаза лейкоцитов (84%), лактатдегидрогеназа (ЛДГ; 82%), α2-макроглобулин (72%), α2-антитрипсин (69%) [25]. В последние годы доказана важная роль в любом воспалительном процессе (в т. ч. при поражении ПЖ) цитокинов, которые выделяются лейкоцитами и поступают в очаг воспаления.
К провоспалительным цитокинам относят интерлейкины (ИЛ) 1, 6, 8, фактор некроза опухоли (ФНО). Их антагонисты – противовоспалительные медиаторы – ИЛ‑10, антагонист рецепторов ИЛ‑1 и др. При дисбалансе про- и противовоспалительных агентов в сторону первых воспаление, в т. ч. при панкреатите, усиливается.
В связи с этим в качестве маркеров воспаления при панкреатитах широко используют уровни провоспалительных ИЛ в крови. Доказано, что через сутки от начала ОП или атаки ХП в крови повышается содержание ИЛ‑6, а через 48 ч – ИЛ‑8, тогда как концентрация ИЛ‑10, напротив, снижается. Предложена т. н. гипотеза второй атаки [23]: повышение уровня провоспалительных цитокинов в крови наблюдается через 1-2 сут после первой атаки – повреждения ПЖ и гиперферментемии. Результат «второй атаки» – септические осложнения панкреатита и его системные проявления (изменения со стороны почек, печени, легких и т. д.).
Неспецифический маркер воспаления – СРБ, содержание которого в крови увеличивается через 72 ч от развития ОП или начала тяжелой атаки ХП. Через 1-3 сут от начала панкреатической атаки повышается уровень фактора активации тромбоцитов (ФАТ), поэтому одним из современных средств лечения панкреатитов является антагонист ФАТ лексипафант. Универсальным маркером инфицирования (в т. ч. инфицирования панкреонекроза при ОП, тяжелой атаке ХП) является возрастание концентрации прокальцитонина в крови при повторных исследованиях >1,8 нг/мл.
Оценка сывороточных маркеров активности воспаления имеет большее значение для оценки тяжести панкреатита, его прогноза, чем для постановки диагноза, т. к. результаты этих тестов неспецифичны [12].
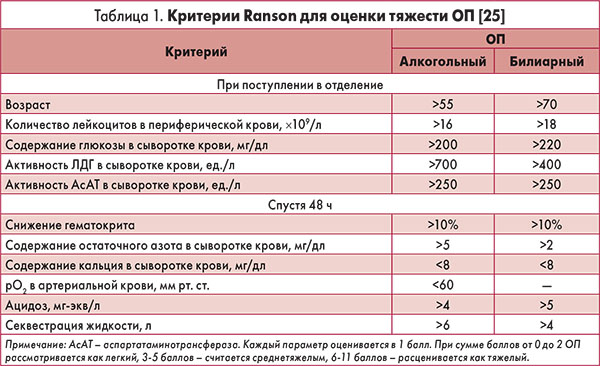
Для оценки тяжести и прогноза ОП используют различные балльные системы, например критерии Ranson (табл. 1), Glasgow, APACHE II и др.
Критерии Glasgow (Imrie) [25]:
• возраст > 55 лет;
• количество лейкоцитов в периферической крови >15×109/л;
• рО2 в артериальной крови 10 ммоль/л (при отсутствии диабета);
• уровень мочевины в крови >16 ммоль/л;
• концентрация кальция в сыворотке крови • содержание белка в сыворотке крови 600 ед./л;
• активность трансаминаз (АсАТ, аланинаминотрансферазы – АлАТ) в сыворотке крови >100 ед./л.
При выявлении у больного ≥3 критериев Glasgow (Imrie) ОП считается тяжелым.
Более динамична, но требует значительного количества сложных исследований классификационная система APACHE II (Acute Physiology And Chronic Health Evaluation). Она предусматривает балльную оценку физиологических показателей (артериального давления, ректальной температуры, сердечного выброса и др.), возраста больного, наличия фоновых хронических заболеваний сердечно-сосудистой системы, органов дыхания, печени, почек. Учет показателей сложен и обычно проводится с помощью компьютерных программ.
Применение лабораторных тестов позволяет уточнить этиологию ОП или ХП, если они связаны с патологией желчных путей (билирубин, щелочная фосфатаза), гиперкальциемией, аутоиммунными заболеваниями (повышение уровня IgG4 в сыворотке крови при аутоиммунном панкреатите), инфекциями (бактерии, вирусы, простейшие, грибы, паразиты).
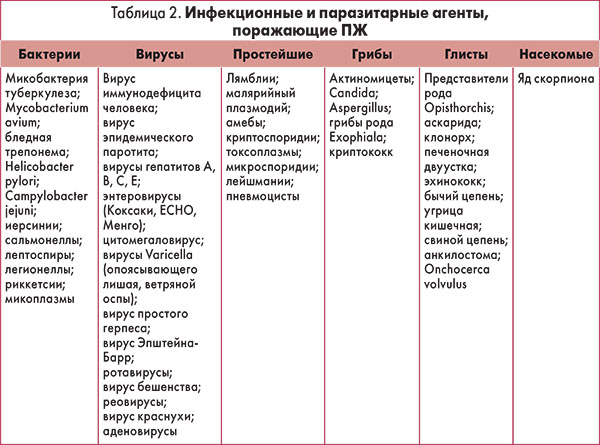
В таблице 2 приведен перечень инфекционных агентов, способных вызвать поражение ПЖ. При наличии соответствующих эпидемиологических и клинических данных необходимо определить лабораторные маркеры этих инфекций.
В последние годы при подозрении на ХП возрастает диагностическое значение генетических исследований, в частности изучение мутаций генов катионного трипсиногена (мутация R122H), ингибитора Казаля типа I (SPINK I; мутация N291), кистозного фиброза (CFTR), мутаций генов метаболизма этанола, выявление наследственного дефицита липопротеинлипазы (приводит к гипертриглицеридемии), дефицита α1-антитрипсина, гемохроматоза [12]. Это связано с тем, что некоторые варианты ХП, которые раньше считали идиопатическими, являются генетически обусловленными [15].
В 1996 г. D. Whitcomb и соавт. разработали генную теорию, объясняющую развитие наследственного панкреатита (НП) и связывающую его с мутацией гена, кодирующего трипсиноген [18]. Была показана ассоциация НП с наследованием специфических маркеров известных локусов хромосом, а также с мутацией гена в длинном плече хромосомы 7 (7q35). При этой мутации происходит замена аргинина на гистидин в положении 117 молекулы трипсиногена – R117H (по новой номенклатуре – R122H) [5, 6]. Доказательство генетической предрасположенности к развитию панкреатита в 5% случаев нашло свое отражение в классификации факторов риска ХП – TIGAR-O. В этой аббревиатуре буква «G» расшифровывается как «genetic» [2, 15]. Важно подчеркнуть, что эта классификация делит пациентов на категории согласно факторам риска, максимально ассоциированным с панкреатитом в каждой конкретной ситуации. Например, пациент с мутацией катионного трипсиногена R122H (вероятность развития панкреатита – 80%, хронизации – 40%), употребляющий алкоголь (изолированное злоупотребление алкоголем без наследственной предрасположенности к панкреатиту сопровождается риском возникновения заболевания <10%), попадет в категорию «Генетический панкреатит», а не в категорию «Токсико-метаболический панкреатит». Действительно, одной генетической предрасположенности, как правило, недостаточно для развития панкреатита – необходим инициирующий внешний фактор (чаще всего – злоупотребление алкоголем, билиарная патология), провоцирующий манифестацию заболевания. Генетическая предрасположенность предопределяет большую вероятность появления того же алкогольного панкреатита [24]. НП одинаково часто наблюдается у мужчин и женщин, не зафиксировано различий в зависимости от расы [6, 7, 16].
Позже были выявлены мутации гена серинпротеазного ингибитора Казаля типа I (SPINK I) у пациентов с идиопатическим ХП, доказана связь с мутацией SPINK I (N291) примерно 50% случаев тропического панкреатита и значительной части таковых идиопатического ХП [14, 17, 20, 21].
Проанализируем подробнее патогенез НП, ассоциированного с мутациями R122H и N291. Молекула трипсина состоит из двух субъединиц, соединенных полипептидной цепью. В положении 117 этой цепи находится аргинин. Между двумя субъединицами трипсина располагается его активный центр, который способен распознавать аргинин и лизин и осуществлять в месте соединения этих аминокислот лизис полипептидной цепи. Именно таким образом трипсин, мезотрипсин и энзим Y инактивируют 80% интрапанкреатических трипсиногена и трипсина [7, 27]. Остальные 20% инактивации интрапанкреатических протеаз обеспечиваются SPINK I. Этот ингибитор представляет собой специфический субстрат для трипсина. SPINK I необратимо связывает серин трипсина с лизином своего активного центра. Важно, что SPINK I синтезируется в количестве, в 20 раз меньшем такового трипсиногена, продуцируемого ПЖ, и может полностью ингибировать трипсин в ткани органа только тогда, когда уровень активности трипсина низкий. В этих случаях SPINK I предотвращает последующую аутоактивацию трипсиногена и блокирует каскад активации панкреатических ферментов и аутолиза ПЖ (рис. 6).
![Рис.6.Протеолитический каскад – основа аутолиза ПЖ [11]](http://health-ua.com/wp-content/uploads/2016/11/61_3.jpg) Рис.6.Протеолитический каскад – основа аутолиза ПЖ [11]
Рис.6.Протеолитический каскад – основа аутолиза ПЖ [11]При интенсивной активации трипсиногена SPINK I не в состоянии его нейтрализовать. В этом случае трипсин и другие трипсиноподобные ферменты, как было сказано выше, лизируют полипептидную цепь, объединяющую 2 субъединицы трипсина, в положении 117, т. е. в месте соединения аргинина и лизина. При мутации катионного трипсиногена R122H аргинин заменяется на гистидин, поэтому трипсин не способен лизировать молекулы трипсиногена и трипсина. Мощности SPINK I в этих случаях не хватает для блокирования аутоактивации трипсиногена, продолжается каскад активации панкреатических ферментов и аутолиз ПЖ [7, 16]. При мутации SPINK I (N291) снижается степень инактивации трипсина, а на фоне воздействия мощного провоцирующего фактора (алкоголь) также развивается НП (рис. 7).
![Рис.7. Интрапанкреатическая активация трипсиногена и генетически детерминированные аномалии механизмов защиты от чрезмерной интраорганной активации этого профермента [11]](http://health-ua.com/wp-content/uploads/2016/11/61-6.jpg) Рис.7. Интрапанкреатическая активация трипсиногена и генетически детерминированные аномалии
Рис.7. Интрапанкреатическая активация трипсиногена и генетически детерминированные аномалиимеханизмов защиты от чрезмерной интраорганной активации этого профермента [11]
НП подразделяется на классический (аутосомно-доминантный, с пенетрантностью 80%; ген катионного трипсиногена PRSS1, R122H, N291) и идиопатический (PRSS1 – A16V, D22G, K23R).