


4 жовтня, 2016
Уникальный механизм действия новых гиполипидемических лекарственных средств
Среди нарушений обмена веществ одно из лидирующих мест занимают нарушения обмена липидов, или дислипидемии. Термин «дислипидемии» (дислипопротеинемии) включает в себя наследственные или приобретенные состояния, характеризующиеся нарушением синтеза, катаболизма, удаления из циркуляции липидов и липопротеинов, что приводит к нарушению их содержания в крови (повышению или понижению). В сравнении с термином «дислипопротеинемия» более широким по охвату нарушений уровня липопротеинов в крови считается термин «гиперлипопротеинемия», который отражает повышенное содержание в крови одного или нескольких классов липопротеинов.
Клиническими проявлениями дислипидемии являются ксантомы – плотные узелки, содержащие холестерин, например, на кисти, подошвах ног, ладонях, коже любого участка тела, особенно спины, а также ксантелазмы – отложение холестерина под кожей век в виде плоских узелков желтого цвета или узелков, не отличающихся по цвету от других участков кожи. При дислипидемии наблюдаются атеросклеротические поражения органов. Атеросклероз – хроническое заболевание, характеризующееся поражением артерий эластического и мышечного типа в виде очаговых отложений липидов и белков в интиме сосудов и реактивной клеточной реакции их стенки, что ведет к сужению просвета сосудов и нарушению физиологических функций пораженных артерий и, как следствие, к расстройствам системы кровообращения.
Сегодня в лечении дислипидемии используется 5 основных классов лекарственных средств (ЛС), применяемых с учетом механизма их действия, эффективности и возможности появления нежелательных эффектов: статины; никотиновая кислота и ее производные; фибраты; секвестранты желчных кислот; антиоксиданты (1).
Среди этих ЛС особого внимания заслуживают статины, появление которых стало возможным благодаря открытию японского ученого Akira Endo, выделившего в 1971 г. из грибов рода Penicillium компонент под названием «компактин», ставший родоначальником статинов. Это соединение обладало способностью снижать уровень холестерина. С тех пор прошло достаточное количество времени, прежде чем на фармацевтическом рынке в 1987 г. появился первый коммерческий статин под названием «ловастатин» (Мевакор), выделенный из гриба рода Aspergillus. Механизм действия статинов реализуется через ингибирование фермента 3-гидрокси-3-метилглютарил-кофермента А редуктазы (ГМГ-КоА-редуктаза), что приводит к уменьшению уровня внутриклеточного холестерина с последующей активацией транскрипции гена рецепторов липопротеинов низкой плотности (р-ЛПНП) и, соответственно, снижению содержания ЛПНП в крови. Статины являются общепризнанным золотым стандартом в лечении гиперлипидемии. За 30 лет их применения они доказали свою эффективность и стали доминирующей группой препаратов, используемых при дислипидемиях.
Однако у каждого пятого человека, принимающего статины, по разным причинам не удается достичь целевых показателей липидов. Некоторые пациенты не переносят статины из-за развития побочных реакций (миалгия, миопатия, миозит, рабдомиолиз, увеличение риска повышения уровня глюкозы в крови и развитие сахарного диабета 2 типа, поражение печени). Кроме того, у значительного количества пациентов с высокой гиперхолестеринемией, в первую очередь семейной гиперхолестеринемией (СГ), даже высоких доз статинов и вспомогательной терапии недостаточно для достижения целевых уровней липидов (2,3). Гиперхолестеринемия – это повышение уровня общего холестерина в 2-2,5 раза по сравнению с нормой за счет ЛПНП. СГ – наиболее распространенное (и очень опасное) наследственное заболевание в мире, без своевременного лечения которого больным угрожают раннее и агрессивное развитие атеросклероза, стеноза аорты, ранние инфаркты, инсульты и наступление внезапной смерти. Как сказал J. Kastelein (4), без лечения у больных с СГ прогноз такой же плохой, как и у больных СПИД(ом). Гетерозиготная форма СГ в общей популяции в большинстве стран встречается довольно часто (от 1:200 до 1:500), в то время как гомозиготная форма проявляется намного реже (1:1 000 000 человек). Считается, что во всем мире больных СГ может быть от 20 до 34 млн, но выявлено заболевание не более чем у 10% из них, а адекватное лечение получают только 5%.
Учитывая важность проведения гиполипидемической терапии для больных с нарушениями липидного обмена, были разработаны ЛС с новыми, уникальными, механизмами действия. Эти ЛС отличаются от известных классов гиполипидемических средств, таких как статины, секвенаторы желчных кислот, ингибиторы абсорбции холестерина.
Ингибитор микросомального триглицерид-переносящего белка
Аполипопротеин В (апоВ) играет ключевую роль в метаболизме липопротеинов. Ген апоВ человека находится на хромосоме 2 и продуцирует с помощью уникального процесса редактирования матричной рибонуклеиновой кислоты (мРНК) 2 физиологические изоформы циркулирующих липопротеинов – апоВ-48, имеющий м. м. 241 кДа и апоВ-100 с м. м. 512 кДа (5). Цифровые обозначения при букве «В» являются условными. Так, максимальная молекулярная масса апоВ принята за 100, а апоВ-48 имеет молекулярную массу, составляющую 48% от максимальной. Сейчас известно до 10 различных т. н. укороченных форм апопротеина В.
ApoB-48 (укороченная форма апоВ-100) продуцируется в кишечнике и играет важную роль при образовании, секреции хиломикронов. Синтез ApoB-100 происходит в печени. Этот белок является фундаментальным структурным компонентом липопротеинов очень низкой плотности (ЛПОНП) и продуктов их метаболизма – липопротеинов промежуточной плотности (ЛППП) и ЛПНП, а также лигандом для р-ЛПНП. Повышение в плазме концентрации апоВ свидетельствует об увеличении количества циркулирующих атерогенных липопротеинов.
Микросомальный триглицерид-переносящий белок – МТТР (Мicrosomal Тriglyceride Тransfer Рrotein) имеет важное значение для сборки и секреции апоВ-содержащих липопротеинов. МТТР впервые был описан в 1984 г. J. Wetterau и D. Zilversmit, выделившими данный белок из микросом печени быка (6). Он состоит из двух субъединиц (М и PDI). В состав М-субъединицы входит 894 аминокислоты (97 кДа). Эта субъединица экспрессируется прежде всего в гепатоцитах и энтероцитах.
Вторая субъединица – широко экспрессируемый многофункциональный белок дисульфид изомеразы – PDI (Protein Disulfide Isomerase) (55кДа) (7). Изомеразная активность PDI не является необходимым условием для функционирования МТТР. Хотя cубъединица PDI и не обладает активностью для переноса липидов, тем не менее она является важным звеном для поддержания активности всего комплекса в целом. Без PDI микросомальный триглицерид-переносящий белок образует нерастворимые агрегаты (8).
МТТР локализован вместе с PDI и ароВ в эндоплазматическом ретикулуме (ЭР) и аппарате Гольджи (АГ), стабилизируя образовавшийся полипептид апоВ и способствуя переносу нейтрального липида из мембраны ЭР в апоВ по принципу челночного механизма. Связывание МТТР с апоВ и перенос липида – независимые процессы. Активность МТР необходима для секреции апоВ-48 и апоВ-100.
МТТР в качестве терапевтической мишени
Исследования, которые проводились в 90-х годах прошлого века, установили, что ингибирование МТТР с использованием фармакологических препаратов приводит к снижению апоВ-содержащих липопротеинов, что дает возможность использовать данный процесс в лечении гиперлипидемии. Впервые в 1998 г. J. Wetterau и соавт. (9) описали небольшую молекулу ингибитора MTТP, она эффективно ингибировала секрецию апоВ-содержащих липопротеинов у грызунов и нормализовала гиперхолестеринемию у гиперлипидемических кроликов. Для этого использовали несколько синтетических ингибиторов MTТP (BMS-201038, СР-346086 и BAY-13-9953), эффективность которых была дозозависимой. Вместе с тем было отмечено, что у лабораторных животных, получавших данные соединения, наблюдалось обратимое накопление жира в печени и энтероцитах. Результаты экспериментов на животных позволяли прогнозировать развитие таких же побочных эффектов, которые могли возникнуть при клинических испытаниях ингибиторов МТТР на человеке. В связи с этим программа исследований была досрочно прекращена из-за развития таких нежелательных явлений, как нарушения со стороны желудочно-кишечного тракта и стеатоз печени (10). Однако спустя несколько лет работа по изучению ингибитора МТТР возобновилась, в результате чего появилось новое ЛС под названием «ломитапид».
Лекарственное средство ломитапид
Биотехнологическая компания Aegerion Pharmaceuticals, Inc. (США) сообщила об одобрении в декабре 2012 г. Управлением по контролю качества пищевых продуктов и лекарственных препаратов (FDA) препарата ломитапид (Lomitapide, Juxtapid). Показанием к применению данного ЛС является гомозиготная СГ – редкое наследственное заболевание, которое сопровождается очень ранним развитием атеросклероза.
Ломитапид – мощный селективный ингибитор МТТР, внутриклеточного липид-переносящего белка, который обнаруживается в просвете эндоплазматического ретикулума и отвечает за связывание и транспортировку отдельных молекул липидов между мембранами. MTТP играет ключевую роль в сборке апоВ-содержащих липопротеинов в печени и кишечнике. Ингибирование МТР приводит к уменьшению синтеза ЛПОНП, ЛППП, ЛПНП и, как результат, – снижению уровня ЛПНП в крови (рис. 1).
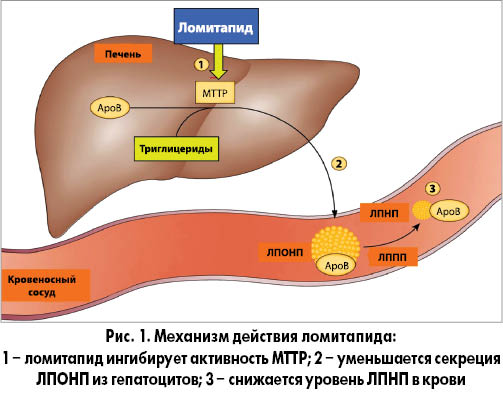
В одном из проводившихся ранее международных клинических исследований было показано, что пациенты с гомозиготной СГ, получавшие ингибитор МТТР, имели значительное снижение липидных показателей по сравнению с исходным уровнем: общий холестерин – ↓58,4%; ЛПНП – ↓50,9%; ЛПОНП – ↓78,7%; триглицериды – ↓65,4%.
Рекомендуемая начальная доза составляет 5 мг 1 раз в день. После 2 нед доза, с учетом приемлемой безопасности и переносимости, может быть увеличена до 10, а затем до 20 и 40 мг. Максимальная рекомендуемая доза – 60 мг.
Антисмысловые олигонуклеотиды
Антисмысловые олигонуклеотиды (АСО) представляют собой синтетические одноцепочечные молекулы дезоксирибонуклеиновой кислоты (ДНК) длиной от 8 до 50 нуклеотидов, которые целиком или частично связываются с рибонуклеиновой кислотой (РНК) и препятствуют дальнейшей трансляции мРНК в белок (11). Подавление биосинтеза белка под действием АСО происходит вследствие того, что мРНК-мишень в составе гибридного ДНК/РНК комплекса с АСО расщепляется внутриклеточной РНКазой Н (12) (рис. 2).
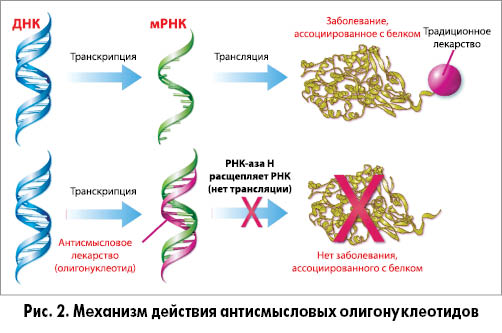
Впервые возможность избирательного подавления продукции определенного белка под действием АСО была продемонстрирована еще в 1978 г. в работе Р. Zamecnik и М. Stephenson (13). Авторы показали, что 13-звенный олигонуклеотид, комплементарный 3'-концевой последовательности РНК вируса саркомы Рауса, ингибирует репликацию вируса in vitro. Эта работа послужила толчком к изучению потенциала АСО в качестве ЛС для лечения вирусных заболеваний, воспалительных процессов, болезней крови, нарушений сердечно-сосудистой системы (14-18).
Ввиду того что природные олигодезоксирибонуклеотиды in vivo подвергаются быстрой деградации под действием нуклеаз, для повышения их стабильности в структуру АСО вносят различные химические модификации (19). Внесение модификаций в структуру олигонуклеотида не только повышает устойчивость АСО к нуклеазам, но и увеличивает эффективность их биологического действия, улучшает гибридизационные свойства и облегчает их захват клетками.
В первом поколении устойчивость к нуклеазам обеспечивалась модификацией фосфодиэфирной связи – замещение кислорода серой (фосфотиоаты), что позволяло повысить устойчивость препаратов к эндонуклеазам, не ухудшая при этом субстратные свойства комплекса относительно нуклеазы Н, однако затруднялось связывание АСО с целевой мРНК. Такие препараты АСО плохо проникали в клетку и имели ограниченный спектр распределения в органах и тканях. Препараты второго поколения были модифицированы по 2'-положению рибозы путем введения 2'-О-метильной, 2'-О-метоксиэтильной группы. Такая модификация позволяла повысить устойчивость к эндонуклеазам и снизить токсичность препарата, но ухудшала субстратные свойства дуплекса с целевой мРНК относительно РНКазы Н. Комбинация двух модификаций позволила получить АСО, обладающие хорошей специфичностью, селективностью и аффинностью к целевой молекуле (20). Химерные АСО, использующие одновременно фосфотиоатные и 2'-О-метоксиэтил или 2'-О-метил-модификации, легли в основу ЛС под названием «мипомерсен».
Лекарственное средство мипомерсен
Мипомерсен (Mipomerse Sodium, Kynamro) производства компании Genzyme Corporation, Isis Pharmaceuticals (США) одобрен FDA в январе 2013 г. Представляет собой синтетический 20-мерный 2'-О-(2-метокси)этил-модифицированный олигонуклеотид, второе поколение одноцепочечного АСО с конкретным комплементарным связыванием кодирующей области мРНК [нуклеотида 3249-3269] В-100 человека. Этот ингибитор имеет 20 нуклеотидов с кодирующей последовательностью 5'-GCCTCAGTCTGCTTCGCACC-3' и является химически стабилизированным через фосфотиоат (21).
В нуклеотидную область мипомерсена включены сахара 2'-дезоксирибозы: 2'-O-метил (2'OMe) и 2'-O-метоксиметил (2'MOE) модифицированной рибозы в 3' и 5' концевых участках. В этом заключается отличие второго поколения АСО от первого, которое легко разрушается эндонуклеазой и экзонуклеазой и оказывает токсическое воздействие на рост и пролиферацию клеток здоровой ткани. Второе поколение АСО мипомерсена значительно эффективнее по сравнению с первым, поскольку обладает большей активностью, имеет повышенную устойчивость к деградации под действием ферментов нуклеаз, более длительный период полураспада и вызывает меньше побочных реакций (27-30).
Aнтисмысловые олигонуклеотиды предназначены для специфического связывания с мРНК апоВ-100 и предотвращения транслокации мРНК для формирования функционального апо-B100 (22-24). Эндогенные РНКазы ферментов, такие как РНКаза Н или Arganaute 2, распознают двойную мРНК апоB-100 и мипоперсена, разрушая тем самым цепь мРНК, в результате чего функциональный белок апоВ-100 не образуется (25,26). К тому же, разрушаясь под действием ферментов, апоВ-100 не экспрессирует из-за неправильного сплайсинга пре-мРНК для зрелой и функциональной мРНК.
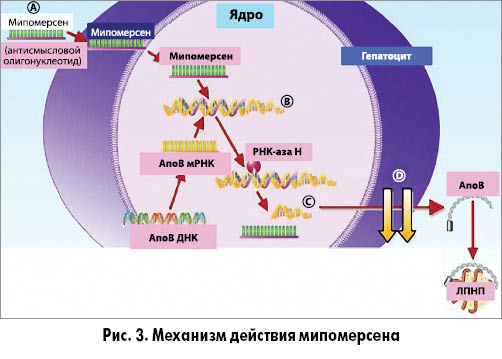
Механизм действия мипомерсена (рис. 3) (31):
А – мипомерсен (одноцепочечный антисмысловой олигонуклеотид);
В – образуется антисмысловая двойная мРНК. После того как мипомерсен достигает гепатоцита, он проникает через мембрану клетки в ядро (механизм не ясен) и связывается с мРНК апоВ с высокой степенью точности;
С – РНКаза Н (неспецифическая эндонуклеаза, которая гидролизует фосфодиэфирные связи РНК при гибридизации с ДНК) расщепляет мРНК. После гибридизации с мРНК-мишенью мипомерсен-мРНК распознается и расщепляется эндогенной РНКазой Н, ферментом, участвующим в репликации/восстановлении ДНК;
D – результатом расщепления апoB мРНК является снижение синтеза апоВ и уменьшение ЛПНП в сыворотке.
В клинических исследованиях было продемонстрировано, что у пациентов, принимающих мипомерсен, среднее снижение липидных параметров (в %) от исходного уровня оставалось неизменным через 104 нед: ЛПНП – ↓27-28%; апоВ – ↓28-31%; липопротеин – ↓17-21%; ЛПВП – ↑3-10%; триглицериды – ↓3-14%.
Мипомерсен в дозе 200 мг вводится подкожно 1 раз в неделю.
Моноклональные антитела, блокирующие фермент пропротеиновую конвертазу субтилизин-кексинового типа 9
В середине 80-х годов ХХ в. американские ученые J. Goldstein и М. Brown получили Нобелевскую премию по медицине и физиологии за открытие рецепторов ЛПНП и установление причин развития СГ. Результаты их исследований убедительно продемонстрировали, что ЛПНП удаляются из кровотока через рецептор-опосредованный путь и что экспрессия новых р-ЛПНП регулируется внутриклеточным содержанием холестерина по механизму отрицательной обратной связи (32). Однако механизмы разрушения уже существующих р-ЛПНП долгое время оставались непонятными. Только в 2003 г. был обнаружен ответственный за этот процесс фермент – пропротеин конвертаза, относящаяся к семейству сериновых протеаз и получившая название «пропротеиновая конвертаза субтилизин-кексинового типа 9» (PCSK9 – от Proprotein Convertase Subtilisin/Kexin type 9). PCSK9 – фермент-гидролаза, продукт гена человека PCSK9. Относится к семейству пропротеиновых конвертаз, которые активируют ферменты, отщепляя от них пептид, ингибирующий их каталитическую активность. PCSK9 играет важную роль в гомеостазе холестерина и является потенциальной мишенью для агентов, снижающих уровень холестерина ЛПНП в крови. Показано, что синтезированная в печени PCSK9 секретируется в кровоток и образует комплекс с р-ЛПНП на поверхности гепатоцита. Не проявляя протеолитической активности, PCSK9 способствует разрушению рецептора после интернализации образовавшегося комплекса в клетку (33,34).
Механизм регуляции PCSK9 экспрессии рецепторов ЛПНП на гепатоцитах
Печень является основным органом, отвечающим за клиренс и катаболизм сывороточных ЛПНП. Гепатоциты регулируют уровень ЛПНП, экспрессируя р-ЛПНП, которые связывают ЛПНП и удаляют их из плазмы. Комплекс ЛПНП/р-ЛПНП интернализуется в гепатоцит в составе клатриновых пузырьков, которые затем сливаются с эндосомами. Кислая среда внутри эндосом способствует диссоциации комплекса ЛПНП/р-ЛПНП. После диссоциации свободные р-ЛПНП повторно возвращаются на поверхность гепатоцита, где они связывают и выводят из кровотока новые частицы ЛПНП. Было показано, что на поверхности гепатоцита происходит кальций-зависимое взаимодействие между каталитическим доменом PCSK9 и повтором А EGFР домена рецептора ЛПНП. После того как ЛПНП связывается с таким рецептором, весь комплекс – ЛПНП/р-ЛПНП/PCSK9 перемещается в клетку в составе клатринового пузырька. В дальнейшем кислая среда эндосомы способствует, с одной стороны,отделению ЛПНП от комплекса, а с другой – повышению аффинности взаимодействия PCSK9 с р-ЛПНП за счет формирования дополнительных ионных связей между про-доменом PCSK9 и р-ЛПНП. В результате PCSK9, не проявляя протеолитической активности, удерживает, как распорка, р-ЛПНП в открытом положении, не давая ему принять закрытую конформацию, необходимую для повторного возвращения на поверхность клетки. Это ведет к тому, что количество экспрессированных на гепатоците рецепторов снижается, соответственно – уменьшается клиренс ЛПНП из сыворотки (35).
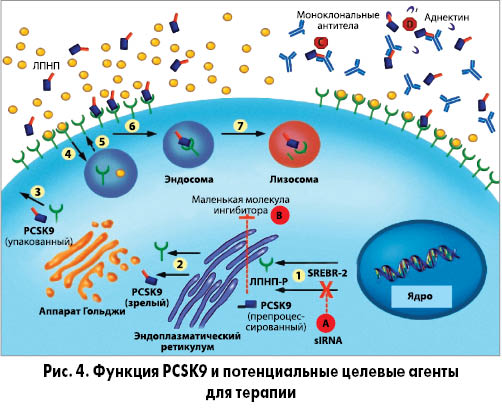
Существует несколько способов влияния на функцию PCSK9 (обозначены буквами в красных восьмиугольниках) (рис. 4):
А – маленькие интерферирующие рибонуклеиновые кислоты (siRNAs) могут блокировать транскрипцию PCSK9 мессенджера рибонуклеиновой кислоты;
В – маленькие молекулярные ингибиторы могут нарушить процессинг PCSK9, что приведет к снижению образования функциональных PCSK9;
С, D – моноклональные антитела и аднектин ингибируют функцию PCSK9.
1 – SREBP-2 (стерол-связывающий регулирующий элемент-2) регулирует в гепатоцитах транскрипцию нескольких липидов и белков, в том числе рецепторов ЛПНП и PCSK9;
2 – PCSK9 превращается в эндоплазматическом ретикулуме в зрелую форму;
3 – упаковывается в аппарате Гольджи (как это делает рецептор ЛПНП) и затем секретируется;
4 – р-ЛПНП связывает циркулирующие ЛПНП;
5 – комплекс перемещается в эндосомы, где ЛПНП могут использоваться для других целей.
р-ЛПНП рециркулирует на клеточной поверхности до 150 раз, удаляя ЛПНП из циркуляции;
6 – PCSK9 регулирует количество р-ЛПНП путем их связывания и интернализации;
7 – разрушение происходит в лизосомах, что не позволяет рецепторам ЛПНП рециркулировать.
Указанные исследования дали понимание того, что снижение активности PCSK9 может стать терапевтической мишенью в лечении пациентов с гиперхолестеринемией, а это позволило бы изолированно снижать уровень ЛПНП. Блокада этого фермента приводит к экспрессии большего количества рецепторов ЛПНП на поверхности печени. В результате захват ЛПНП печенью увеличивается, их содержание в крови уменьшается. Блокирование PCSK9 является инновационным механизмом снижения ЛПНП. Из всех потенциальных ингибиторов PCSK9 для практического использования были выбраны моноклональные антитела человека (ЛС алирокумаб).
Лекарственное средство алирокумаб
Алирокумаб (Alirocumab, Praluent), совместная разработка фармацевтической компании Sanofi (Франция) и биофармацевтической компании Regeneron Pharmaceuticals (США), одобрен FDA в октябре 2015 г. Препарат представляет собой моноклональные антитела человека (изотип IgG1), которые связываются с PCSK9 с высокой аффинностью и специфичностью и производятся с использованием технологии рекомбинантной ДНК в суспензионной культуре клеток яичников китайского хомячка. Алирокумаб состоит из двух дисульфид-связанных тяжелых цепей, каждая из которых ковалентно связана с легкими каппа-цепями. Единственный N-связанный сайт гликозилирования локализован в каждой тяжелой цепи на CH2 домене Fc константной области молекулы. Вариабельные домены тяжелой и легкой цепей в совокупности образуют сайт связывания PCSK9. Приблизительная молекулярная масса алирокумаба составляет 146 кДа.
Механизм действия алирокумаба показан на рисунке 5.
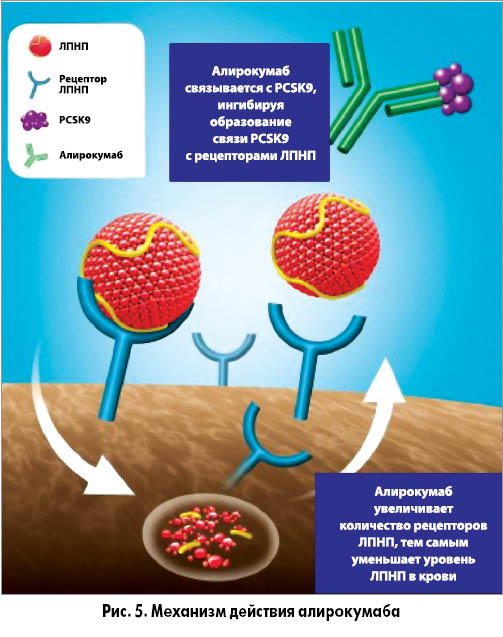
Рекомендуемая начальная доза алирокумаба составляет 75 мг. Вводят подкожно 1 раз в 2 нед. Если ответ ЛПНП является неадекватным, доза может быть увеличена до 150 мг.
Появление на мировом фармацевтическом рынке новых гиполипидемических препаратов с уникальным механизмом действия, удобным режимом дозирования и высокой эффективностью предоставляет врачам-клиницистам более широкие возможности в выборе средств терапии пациентов с дислипидемией.
Список литературы находится в редакции.
Медична газета «Здоров’я України» № 17 (390), вересень 2016 p.