


2 червня, 2023
Характеристика та визначення рівня фактора VIII при порушенні системи згортання крові
Фактор зсідання крові VIII (або антигемофільний фактор А) являє собою глікопротеїн, який синтезується здебільшого в гепатоцитах, а також у нирках, ендотеліальних клітинах та лімфоїдній тканині. Це один із найбільших факторів згортання (містить 2332 амінокислоти (а/к), молекулярна маса становить 293 кДа), наявний у кровотоці в комплексі з фактором фон Віллебранда (vWF), який захищає його від передчасного протеолізу [1-3]. Період напіврозпаду фактора VIII складає ≈12 год, концентрація в плазмі – 0,2 мкг/мл. Активна форма (фактор VIIIa) є неферментативним кофактором протромбіназно-теназного комплексу в системі внутрішнього згортання крові. Водночас відбувається прискорення активації фактора X, спричинене активованим фактором IX (фактор IXa), за наявності фосфоліпідів та іонів кальцію. Ген фактора VІІІ розташований на Х-хромосомі (Xq28). Мутація в ньому зумовлює вроджене порушення згортання крові, тобто гемофілію А [4, 5]. Ця мутація зустрічається майже винятково в чоловічих статевих клітинах. Гемофілія А діагностується в 1 із 5000 новонароджених чоловічої статі.
Біохімічна характеристика фактора VIII
Фактор VIII згортання крові людини кодується геном, що складається зі 186 000 пар основ; містить 26 екзонів і 25 інтронів, при цьому 14-й екзон є найбільшим. Незвичайність гена – це наявність 2 додаткових генів, відомих як гени F8A та F8B в інтроні 22 (IVS22), функції яких наразі невідомі. Фактор VIII має 6 доменів (А1-А2-В-А3-С1-С2) [6] (рис. 1).
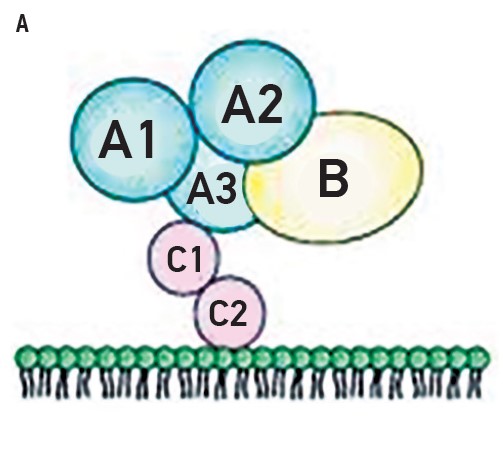
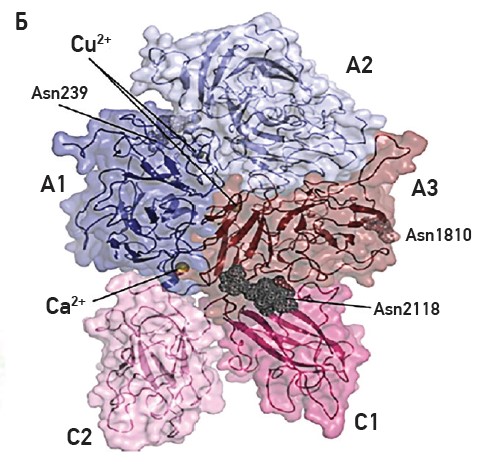
Рис. 1. А – А1, А2, В, А3, С1, С2 – домени, що утворюють фактор згортання крові VIII; Б – тривимірна структура фактора коагуляції VIII з видаленим доменом B
У крові під впливом протеолітичних процесів (фуринова протеаза) цей білок розподіляється на 2 ланцюги: важкий – 200 кДа (А1-А2-В) та легкий – 80 кДа (A3-C1-C2). Ланцюги пов’язані між собою ковалентним зв’язком. Обмежений протеоліз В-ланцюга спричиняє гетерогенну популяцію форми активного фактора VIII різної молекулярної маси (від 90 до 200 кДа). Найменша із форм важкого ланцюга (90 кДа), що генеруються, разом із легким ланцюгом (80 кДа) утворюють активну форму коагуляційного фактора VIII. Слід зазначити, що сам фактор VIII неактивний чи мінімально активний як кофактор процесу згортання крові. Активація його як кофактора відбувається тільки після протеолітичного розщеплення [7, 8].
А-домени
Розглянемо А-домени фактора VIII, гомологічні фактори зсідання крові V і церулоплазміну. У структурі фактора VIII є 3 домени А (А1-336 а/к, А2-337 а/к, А3-329 а/к). Дисульфідні зв’язки розташовані вздовж нижньої поверхні білка [9]. Обидва домени (A1 та A3) містять 1 атом міді [10]. Основний епітоп антитіл знаходиться в домені А2 (сайт 484-508 а/к), ще 1 розташований між амінокислотами 558 і 565, які впливають на фактор IXa та, ймовірно, є основою каталітичної активації кофакторів і ферментативних реакцій [11]. Домен A2 може дисоціювати найлегше в активній формі фактора VIII. Кінець пептидної послідовності домену А2 (амінокислоти з 1810 по 1818) утворює специфічний сайт зв’язування з IXa [12]. Специфічне зв’язування із сайтом фактора X відбувається на кінці домену A1 (амінокислоти із 337 по 372) [13]. Ділянка для зв’язування активного протеїну С (АРС), інгібітора процесу коагуляції, розташована на сайті домену А1 (Arg336) та домену А2 (Arg562) [14].
Домен B
Центральний домен В є найбільшим (40% від маси фактора VIII) з усіх доменів і значно глікозильованим [15]. Цей домен впливає на активність форми VIII у процесі коагуляції. Було показано, що природний або рекомбінантний фактор VIII з видаленим доменом B має зіставні чи навіть вищі активності. Його функція остаточно не вивчена.
С-домени
У структурі фактора VIII є 2 С-домени (С1 та С2). Домени C1 і C2 містять 153 та 160 амінокислот відповідно. Кристалічна структура домену С2 складається з β-сендвіча, що має внутрішню доменну структуру, а також приєднані β-петлі, які утворюють гідрофобну поверхню. На вершині першої β-шпилькової структури є амінокислоти Met2199 та Phe2200. Ці 2 гідрофобні амінокислоти приєднані до сусідніх петель. Вони позначають середину поверхні домену C2, що відповідає за фосфоліпідний зв’язок до фактора VIII. На протилежному домені C2 збоку знаходиться сайт зв’язування доменів C1 та A1. Хоча домен С1 не відіграє специфічної ролі у функціонуванні фактора VIII, нещодавні дослідження продемонстрували, що він впливає на vWF [16].
Механізм активації фактора VIII
Фактор зсідання крові VIII протеолітично активується тромбіном. Активація відбувається в результаті розщеплення у важкому ланцюзі: Arg372 (домени A1-A2), Arg740 (домени А2-В) і розщеплення в легкому ланцюзі: Arg1689 (домени B-A3). Активна форма фактора VIII (а саме фактора VIIIa) являє собою тример, що складається з А1 (1-372 а/к), А2 (373-740 а/к) і пов’язаних разом А3 / домени С1-С2 (1690-2332 а/к). Активний фактор VIII не містить В-домену. Функція фактора VIIIa в коагуляційному каскаді полягає у прискоренні активації фактора X за наявності фактора IXa, фосфоліпідів та іонів кальцію [17].
Після індукованої тромбіном активації фактора VIII фактор VIIIa зв’язується з поверхнею фосфоліпідів і взаємодіє з фактором IXa. Фактор VIIIa прискорює активацію фактора X у ≈105 разів. Інактивація фактора VIIIa є результатом розщеплення доменів А1 та А2 в специфічних сайтах Arg336 і Arg562 APC [18].
Роль фактора VIII у згортанні крові
Ініціювальною подією утворення тромбіну є зовнішній шлях системи гемостазу, який запускається мембранозв’язаним тканинним фактором (TF), що вивільняється після ушкодження судин, щоб активувати фактор VII (фактор VIIа) [19]. У фазі ініціації коагуляції комплекс TF / фактор VIIa (зовнішній теназний комплекс) каталізує активацію як фактора IX, так і фактора X, причому останній є ефективнішим субстратом [20]. Спочатку утворений мембранозв’язаний фактор Xa перетворює незначну кількість протромбіну на тромбін, який, своєю чергою, активує тромбоцити, vWF, фактор VIII [21-24]. Фактор IXa, який генерується комплексом TF / фактор VIIa, з’єднується із фактором VIIIa на мембрані активованих тромбоцитів для створення внутрішнього теназного комплексу, основного активатора фактора X. Згодом фактор Xa поєднується із фактором Va на активованих мембранних рецепторах тромбоцитів, що робить його каталізатором протромбінази, яка перетворює протромбін на тромбін. У каталітичній активації протромбіну протромбіназа є у 300 000 разів активнішою за фактор Xa [25]. Протягом усього періоду як у фазі ініціації, так і у фазі розповсюдження прокоагулянтна активність пригнічується утворенням ферментного стехіометричного комплексу інгібітором (інактивація факторів Va і VIIa активованим протеїном С). У міру того, як коагуляція переходить від фази ініціації до фази поширення головний генератор фактора Xa зміщується від TF / фактора VIIa до комплексу фактор VIIIa / фактор IXa. Комплекс фактор VIIIa / фактор IXa не зазнає інактивації інгібітором шляху TF.
Теназний (фактори IXa, VIIIa, X) та протромбіназний комплекси (фактори Xa, Va, протромбін) збираються разом на поверхні мембран активованих тромбоцитів за наявності іонів кальцію. Фактор Ха та тромбін продукуються теназою та протромбіназним комплексом. Зв’язування факторів IXa, X/Xa та протромбіну із тромбоцитами частково здійснюється через Gla-домени в цих білках. Тромбін не містить Gla-доменів і вивільняється з мембрани, після чого розщеплює фібриноген та інші субстрати. Коли фактор VIII відсутній чи є його брак (як у разі гемофілії А), внутрішній теназний комплекс не збирається, посилення генерації фактора Xa немає і кровотеча не припиняється [26].
Отже, фактор VIII має центральну роль у поширенні гемостатичної відповіді. Циркулювальний фактор VIII приєднується до vWF, який стабілізує фактор VIII та зв’язується із субендотеліальним матриксом, опосередковуючи адгезію тромбоцитів у місцях ушкодження. Друга роль фактора VIII у системі згортання крові полягає у регуляції vWF. Фактор VIII стабілізує мультимери vWF і робить їх сприйнятливішими до деградації металопротеазою ADAMTS‑13 (дезінтегрин та металопротеїназа), тоді як фактор VIIIa, не пов’язаний з vWF, не має такого ефекту [27, 28].
Мутації гена фактора VІІІ при гемофілії А
Гемофілія А є рецесивним, зчепленим із Х-хромосомою, вродженим порушенням згортання крові в результаті зниження або відсутності фактора VIII. Приблизно 340 000 чоловіків у всьому світі страждають на цю коагулопатію. З них 70% мають тяжку форму захворювання, що визначається як активність фактора VIII у плазмі <1% [29-31].
Гемофілія А спричиняється широким спектром мутацій у гені фактора VIII, які зазвичай зустрічаються в чоловічих статевих клітинах. Гемофілія А включає 5415 мутацій фактора VIII, з яких 2158 є унікальними [32]. Мутації фактора VIII були виявлені за допомогою різних методів скринінгу; найчастіше це секвенування всієї ділянки екзонів (наприклад, фланкувальні сайти сплайсингу), які ідентифікують унікальні мутації у більшості пацієнтів [33, 34]. Скринінг не надає змоги визначити конкретну мутацію у незначного відсотка пацієнтів, деякі з яких мають унікальні інверсії, аберації мРНК або ознаки мутацій глибоко в інтронах, що здатні впливати на сплайсинг.
Типи мутацій фактора VIII та його значимість
Приблизно 45% усіх випадків тяжкої гемофілії А припадають на зумовлену інверсією гомологічну рекомбінацію інтрона 22 і споріднені послідовності поза геном фактора VIII [35-37]. Незначні делеції або вставки, міссенс-мутації та безглузді мутації є наступними найпоширенішими мутаціями фактора VIII, що відбуваються у 16, 15 та 14% пацієнтів із тяжким дефіцитом фактора VIII. Іншими мутаціями, що менше ідентифікуються, є великі делеції (5%), мутації сайту сплайсингу (3,5%) та інверсії інтрона-1 (3%). Інверсії, вставки, делеції та безглузді мутації спричиняють гемофілію А, оскільки мРНК є аберантною та рамка зчитування ДНК змінюється. Це означає, що фактор VIII не може бути синтезований. Навпаки, при міссенс-мутаціях зазвичай утворюється дисфункціональний білок зі зниженою активністю згортання крові, хоча вони можуть зумовити нейтральні мутації або поліморфізми.
Тяжкість кровотечі за гемофілії А
Тяжкість кровотечі при гемофілії А класифікується залежно від активності фактора VIII (табл. 1).
|
Таблиця 1. Класифікація тяжкості кровотеч у разі гемофілії А |
|
|
Фактор VIII (активність) |
Класифікація тяжкості кровотечі |
|
<0,01 МО/мл (<1% від норми) |
Тяжка |
|
0,01-0,05 МО/мл (1-5%) |
Помірна |
|
>0,05-<0,40 МО/мл (5-40%) |
Легка |
Однак тяжкість кровотечі залежно від активності фактора VIII не має прямолінійного характеру, адже насправді є пацієнти з рівнем фактора VIII <1%, які мають дуже незначні спонтанні кровотечі (або ж вони взагалі відсутні). З іншого боку, спонтанні та клінічно тяжкі кровотечі мають деякі хворі з 1-5% активністю фактора VIII. Оскільки в пацієнтів із хворобою фон Віллебранда також спостерігається зниження активності фактора VIII, достовірний діагноз гемофілії А потребує визначення антигена vWF або активності кофактора ристоцетину. Один рідкісний варіант (а саме тип 2N хвороби фон Віллебранда, за якої vWF не пов’язується із фактором VIII) може бути помилково діагностований (автосомна!) як гемофілія А, тому в цьому випадку слід провести додаткові обов’язкові дослідження [38, 39]. У хворих із тяжкою формою гемофілії А клінічно зазвичай проявляються крововиливом у суглоби та м’язи, а також тривалими й небезпечними кровотечами після травм і операцій, що може зумовити інвалідність. За помірної або легкої гемофілії епізоди кровотечі є рідкіснішими та з’являються зазвичай через травму чи операцію [40].
Методи визначення фактора VIII
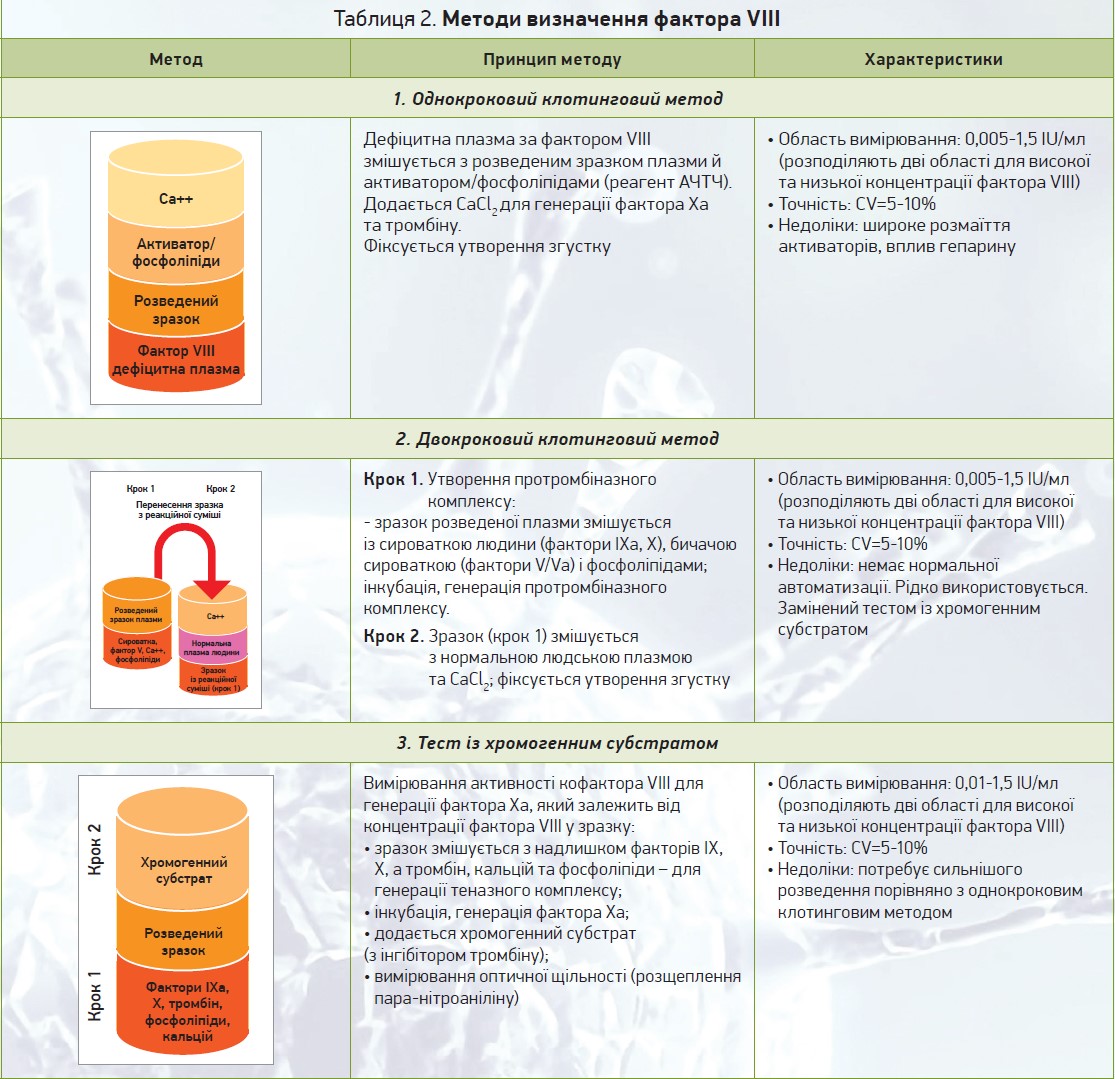
Наразі доступні 3 методи для вимірювання фактора VIII у біологічних зразках:
- однокроковий клотинговий (OSA, One-Stage Assay);
- двокроковий (TSA, Two-Stage Assay);
- аналіз хромогенного субстрату (CSA, Chromogenic Substrate Assay) [41-44] (табл. 2).
Використання плазми, дефіцитної за фактором VIII
Більшість наявних у продажу субстратів плазми (ліофілізованих або заморожених) готують шляхом селективної імуноадсорбції фактора VIII з об’єднаної нормальної плазми людини. Дефіцитну плазму також одержують від пацієнтів із вродженим дефіцитом фактора VIII. Однак не в усіх суб’єктів із вродженим дефіцитом об’єднана плазма може бути повністю позбавлена фактора VIII.
Оскільки плазма з дефіцитом фактора VIII використовується в нерозбавленому вигляді та змішується з рівними частинами щодо високих розведень плазми пацієнта, наявність навіть незначних слідів фактора VIII може вплинути на загальну концентрацію фактора VIII у тесті. Саме тому дефіцитна (субстратна) плазма має бути повністю позбавлена фактора VIII.
Однокроковий клотинговий метод
Однокроковий метод аналізу фактора згортання VIII заснований на вимірюванні ступеня корекції активованого часткового тромбопластинового часу (АЧТЧ) із використанням розведеної плазми пацієнта та фактора VIII дефіцитної плазми. Цей аналіз відображає внутрішній шлях системи згортання крові, в якому реакція ініціюється контактним активатором (наприклад, елаговою кислотою, каоліном, діоксидом кремнію, поліфенолом) із синтетичним фосфоліпідом, що використовується для заміни фосфоліпіду, отриманого із тромбоцитів, оскільки аналізи проводяться із застосуванням бідної на тромбоцити плазми. Завдяки своїй простоті, доступності реагентів, автоматизації та низькій вартості порівняно із хромогенним тестом однокроковий метод використовується в переважній більшості клінічних лабораторій у світі [45-48].
Двокроковий клотинговий метод
Складається із 2 окремих реакцій, безпосередньо пов’язаних з кількістю фактора VIII, що наявний у плазмі пацієнта. На першому етапі для створення протромбіназної активності зразок пацієнта, що містить фактор VIII, додають до сироватки людини (містить активований фактор IX і X) + бича сироватка, абсорбована солями барію (джерело фактора V/Va) + іони кальцію + фосфоліпіди. На другому етапі протромбіназа, що утворюється на першому етапі, перетворює протромбін на тромбін (субстратна плазма являє собою нормальну об’єднану плазму людини), після чого визначається швидкість перетворення фібриногену на фібрин, яка вимірюється як кінцева точка утворення згустку. Оскільки час згортання крові залежатиме від рівня фактора Ха в зразку, отриманому на першому етапі, а рівень фактора Ха є пропорційним концентрації фактора VIII у плазмі пацієнта, активність фактора VIII можна отримати з показників зсідання крові на другому етапі.
Аналіз фактора VIII з використанням хромогенного субстрату
Є аналогічним двоетапному аналізу фактора VIII. Включає стадію інкубації для отримання фактора Ха та другу стадію для визначення кількості фактора Ха, що продукується. В цьому випадку кількість фактора Ха вимірюється за його впливом на конкретний хромогенний субстрат. Оскільки інтенсивність фарбування є прямо пропорційною кількості фактора Xa, яка, своєю чергою, прямо пропорційна кількості фактора VIII, рівні фактора VIII можуть змінюватися. Розраховується за поглинанням зразка за певної довжини хвилі (оптимальна довжина хвилі поглинання для хромофора, отриманого розщепленням хромогена фактора Xa, зазвичай складає 405 нм) (табл. 3).
|
Таблиця 3. Компоненти, необхідні для визначення фактора VIII з використанням хромогенного методу |
|
|
Компонент |
Склад |
|
«Реагентний коктейль» для генерації фактора Xa |
Містить фактори IXa, X у надлишку, тромбін (фактор IIa), джерело іонів кальцію та фосфоліпідів |
|
Хромогенний субстрат |
Може також містити інгібітор тромбіну, щоб зупинити утворення Xa при додаванні хромогена |
|
Плазма пацієнта |
Бідна на тромбоцити плазма |
Процедура визначення фактора VІІІ хромогенним методом
Плазму пацієнта інкубують із сумішшю реагентів за температури 37 °C. Тромбін у «коктейлі» активує фактор VIII і перетворює його на активний фактор VIIIa, який за наявності Ca2+ та фосфоліпідів діє як кофактор фактора IXa для переходу фактора X в активний фактор Xa. Концентрація фактора VIII є стадією, що обмежує швидкість. За додавання хромогенного субстрату фактор Ха гідролізує хромогенний субстрат, вимірюється поглинання отриманого продукту (зазвичай пара-нітроаніліну, що вивільняється ферментативно) та порівнюється з еталонною кривою для отримання рівня фактора VIII (рис. 2).
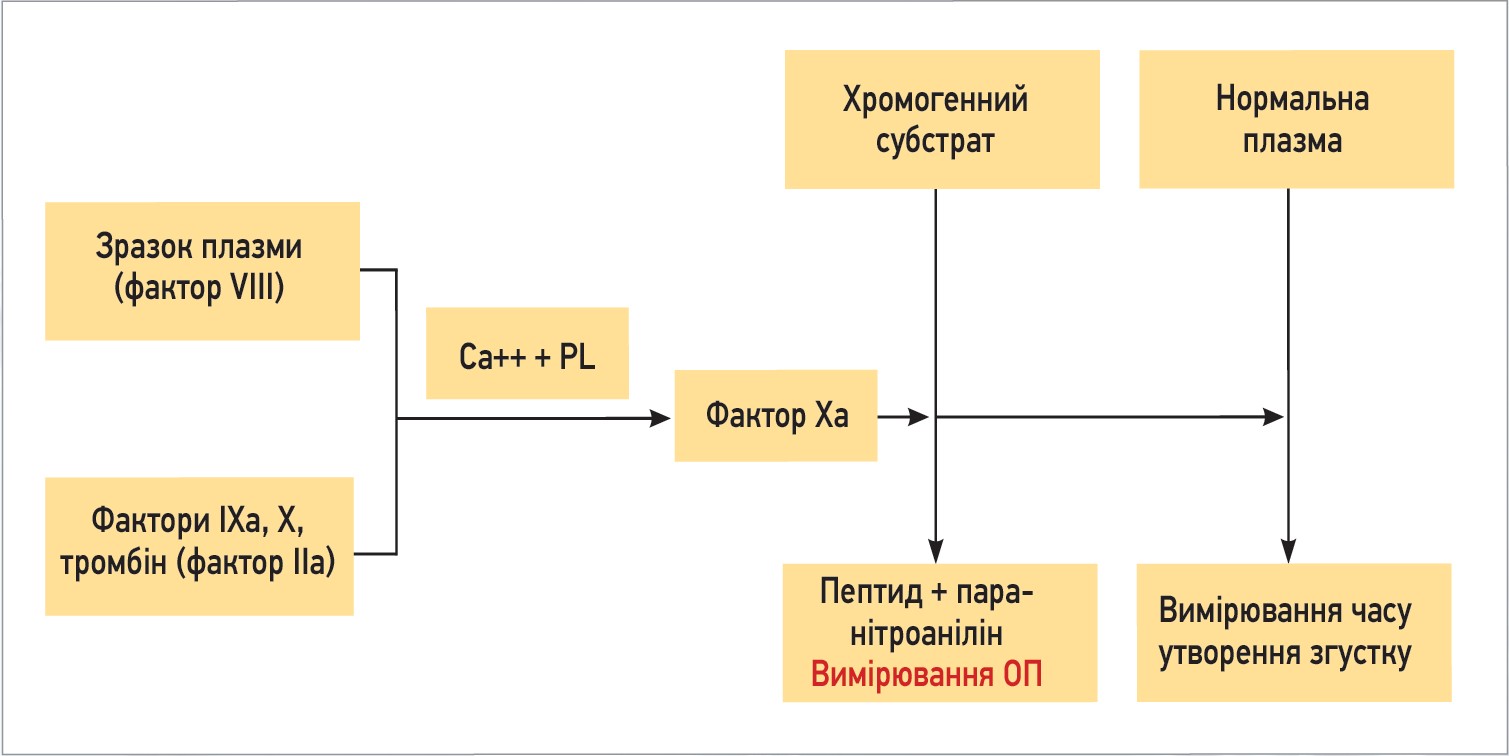
Рис. 2. Принцип аналізу фактора VІІІ з використанням хромогенного субстрату
Деякі особливості визначення фактора VIII
- Якщо дефіцит фактора VIII є новою та несподіваною знахідкою, слід перевірити рівні vWF і фактора V. У цьому немає потреби, якщо очікується виявлення дефіциту фактора VIII (наприклад, у людини з гемофілією А).
- Зазначається збільшення кількості випадків, коли пацієнти з дефіцитом фактора VIII мають невідповідні рівні при вимірюванні з використанням одноетапного аналізу порівняно із двоетапним або хромогенним аналізом. Фенотип кровотечі краще корелює з двоетапним або хромогенним аналізом фактора VIII, тому важливо, щоб у всіх осіб із підозрою на порушення згортання крові було проведено як одноетапний, так і хромогенний аналіз фактора VIII.
- У здорової людини можуть продукуватися автоантитіла до фактора VIII, що спричиняє появу набутої гемофілії А. Такі антитіла також виявляються в пацієнтів із низкою імунологічних порушень, наприклад, гемофілією, ревматоїдним артритом.
- Низький рівень фактора VIII також може бути виявлений за хвороби фон Віллебранда та при набутому синдромі фон Віллебранда.
- Фактор VIII є білком гострої фази; його рівні можуть бути високими в людей, які перебувають у стані стресу, включаючи вагітність. Це може зумовити скорочення АЧТЧ.
- Концентрація фактора VIII є основним фактором, що визначає значення АЧТЧ. Низькі рівні фактора VIII подовжують АЧТЧ, а високі рівні фактора VIII, навпаки, скорочують АЧТЧ.
- У жінок із низьким рівнем фактора VIII або IX та відсутністю відповідного сімейного анамнезу слід установити каріотип, тобто необхідно враховувати синдром Тернера.
Висновки
Фактор VIII є коферментом, критично важливим для прискорення генерації фактора Ха, а згодом – тромбіну. Отже, фактор VIII має центральну роль у поширенні гемостатичної відповіді.
Визначення концентрації фактора VIII використовується для:
- лікування пацієнтів із гемофілією А чи хворобою Віллебранда;
- моніторингу хворих, які отримують замісну терапію фактора VIII;
- оцінки ризику тромбозу. Стійкі рівні активності фактора VIII >150% зумовлюють 5-6-кратне підвищення ризику тромбозу глибоких вен (ТГВ) і рецидивувального ТГВ, ніж рівні активності фактора VIII <100%;
- документування гострофазової відповіді.
Список літератури знаходиться в редакції.
Медична газета «Здоров’я України 21 сторіччя» № 8 (544), 2023 р