


6 серпня, 2024
Хвороба Гоше. Клінічна настанова, заснована на доказах
Хвороба Гоше (ХГ) – це спадкове панетнічне захворювання, яке в більшості випадків починається в дитинстві та зумовлене дефіцитом активності лізосомального ферменту глюкоцереброзидази (кислої β-глюкозидази). У дітей із клінічними проявами перебіг захворювання зазвичай тяжкий, спостерігають затримку росту і статевого дозрівання, ранній розвиток остеопенії, значну спленомегалію, гепатомегалію, тромбоцитопенію, анемію, сильний біль у кістках, гострі кісткові кризи, а також переломи. Дітям із симптомами ХГ необхідно призначати ферментну замісну терапію (ФЗТ), що допоможе уникнути виснажливого і часто незворотного прогресування патології, а також дасть можливість вести нормальний спосіб життя хворим із ненейропатичною формою захворювання.
Ця клінічна настанова є адаптованою для системи охорони здоров’я України версією клінічних рекомендацій Revised recommendations for the management of Gaucher disease in children. European Journal of Pediatrics (2013), що були обрані робочою групою як приклад найліпшої практики надання медичної допомоги пацієнтам із ХГ, і ґрунтується на даних доказової медицини стосовно ефективності і безпеки медичних утручань, фармакотерапії та організаційних принципів її надання.
Вступ
Revised recommendations for the management of Gaucher disease in children. European Journal of Pediatrics (2013)
Поширеність ХГ у світі становить приблизно 1:40 000-1:60 000. Серед ашкеназьких євреїв це одна з найпоширеніших генетичних патологій із частотою випадків серед носіїв 1:17. Успадковується за аутосомно-рецесивним типом. Причиною є дефіцит лізосомального ферменту глюкоцереброзидази (кислої β-глюкозидази), що призводить до накопичення субстрату глюкоцереброзиду (глюкозилцераміду) в лізосомах макрофагів та інших клітин, наприклад остеобластів. Заповнені ліпідами «клітини Гоше» акумулюються в різних тканинах і органах, особливо в селезінці, печінці, кістковому мозку (рис. 1, а), легенях та мозку.
Хоча ХГ і проявляється в рамках певного спектра симптомів, загальноприйнятою є клінічна класифікація з поділом захворювання на три типи: тип І (ненейропатичний, Каталог фенотипових маркерів людини (МІМ) 230 800), тип ІІ (гострий нейропатичний, МІМ 230 900) і тип ІІІ (хронічний нейропатичний, МІМ 23 100). В Європі, Канаді та США найпоширенішою (94%) формою ХГ є тип І, що характеризується відсутністю первинного залучення в патологічний процес центральної нервової системи. В інших країнах світу, таких як Єгипет, Японія, Швеція, Польща, нейропатичні форми можуть бути поширенішими, ніж ХГ типу І.
Ознаки та симптоми ненейропатичної форми ХГ у дітей: спленомегалія, гепатомегалія, тромбоцитопенія, анемія, відставання в розвитку, затримка росту і статевого дозрівання, остеопенія, сильні болі в кістках (гострі кісткові кризи) та переломи. При ненейропатичній формі ХГ, яку часто невірно описують як патологію дорослого віку, у більшості пацієнтів симптоми проявляються в дитинстві, а діагностують захворювання до досягнення 20-річного віку. Ранній початок захворювання асоціюється з тяжчим перебігом і високим ризиком ускладнень. У 75% нелікованих дітей із симптомами хвороби спостерігають субоптимальний розвиток організму, 34-42% із них мають зріст на рівні ≤5 перцентилів. На момент встановлення діагнозу в більш як 80% дітей виявляють спленомегалію, гепатомегалію та патологію кісток. У дітей, які не отримують лікування, середній об’єм селезінки більш ніж у 20 разів перевищує норму, а середній об’єм печінки – удвічі більший за норму для відповідного віку і ваги. У приблизно 60% підлітків із нелікованою ХГ спостерігають затримку статевого дозрівання, що може призвести до значних негативних психологічних наслідків. Анемія і легка або помірна тромбоцитопенія, асоційовані з гіперспленізмом, наявні приблизно в 40% дітей. Анемія є однією з основних причин втоми, а тромбоцитопенія збільшує ймовірність кровотеч (у тому числі частих носових) і синців.
Патологія кісток зумовлена накопиченням клітин Гоше в кістковому мозку, глюкоцереброзиду в остеобластах та дією цитокінів, що їх виробляють ці клітини. Приблизно 80% дітей із ХГ на момент встановлення діагнозу мають щонайменше одну кісткову патологію. На рентгенологічному знімку дистального відділу стегнової кістки можуть спостерігатися характерні колбоподібні деформації Ерленмейєра (рис. 1, б). Деякі діти страждають на хронічний біль у кістках і/або кісткові кризи (гострий початок, тривалі напади болю, що спочатку має тупий характер, надалі стає нестерпним; зазвичай передує появі остеонекрозу та переломів). Мінеральна щільність кісткової тканини (МЩКТ), маса і товщина кортикального шару зазвичай аномально низькі.
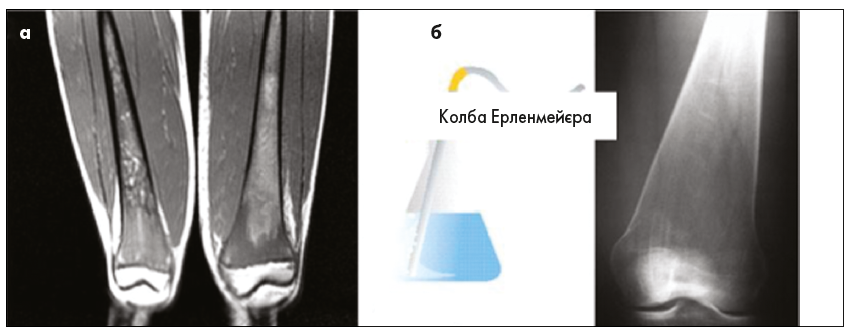
Рис. 1. Інфільтрація кісткового мозку на МРТ-зображенні (а); колбоподібні деформації Ерленмейєра на рентгенологічному знімку (б)
Патогенез ХГ ІІ і ІІІ типів подібний, він відрізняється переважно за ступенем неврологічної дегенерації – швидкий/гострий перебіг характерний для типу ІІ, хронічний – для типу ІІІ. Гостра нейропатична ХГ (ІІ тип) спостерігається дуже рідко (1:500 000) та є панетнічною. Хворі діти можуть бути здоровими при народженні, але до досягнення 2-річного віку в них проявляються системні та неврологічні ознаки патології (часто набагато раніше), які стрімко посилюються. Більшість дітей помирає в ранньому дитинстві, частина – у внутрішньоутробному періоді, інші – упродовж декількох років. ХГ ІІІ типу проявляється в ранньому дитинстві, часто має симптоми, подібні до ХГ І типу, та повільно прогресуючий перебіг, що часто призводить до летального наслідку на 2-3-му десятилітті життя. Такі неврологічні ознаки, як горизонтальна окуломоторна апраксія, порушення погляду по вертикальній лінії, аномально повільне стеження за об’єктом та конвергентна косоокість, зазвичай проявляються в ранньому дитинстві, але можуть і пізніше. Для ХГ типу ІІІ було розроблено шкалу оцінки тяжкості захворювання і виділено щонайменше три підтипи. У пацієнтів із типом ІІІа спостерігають прогресуючу неврологічну патологію з міоклонусом та деменцією. Хворі на тип ІІІb мають значні вісцеральні та скелетні ураження, подібні до симптомів тяжкого типу І, з неврологічними ознаками (переважно горизонтальний над’ядерний параліч погляду), що можуть з’являтися до або після появи соматичних проявів і симптомів. Кісткова патологія зазвичай представлена у вигляді деформацій грудної клітки та вираженого кіфосколіозу без супутнього болю в кістках чи кісткових кризів. Тип ІІІс ХГ характеризується різноманітними неврологічними ознаками (зокрема, гідроцефалією), помутнінням рогівки, кальцифікацією аорти і серцевих клапанів, що стає причиною застійної серцевої недостатності та аритмій.
Призначення ФЗТ дітям із ХГ типів І чи ІІІ і наявністю симптомів може запобігти розвитку серйозних, незворотних соматичних ускладнень (але не неврологічних), які призводять до інвалідизації хворих.
Рекомендації з діагностики та ведення дітей із ненейропатичною формою ХГ були оновлені міжнародними експертами з ХГ кількох медичних спеціальностей, розроблені The Belgian Working Group on Gaucher Disease, Guidelines for diagnosis, treatment and monitoring of Gaucher’s disease (2016).
Діагностика хвороби Гоше
Revised recommendations for the management of Gaucher disease in children. Еuropean Journal of Pediatrics (2013)
Діти з наявністю симптомів
Діагноз ХГ у дітей із наявністю симптомів встановлюють на основі історії хвороби, даних медичного огляду і лабораторних тестів, підтверджених результатами ферментного і/або генетичного аналізу.
В історії хвороби мають бути здуття і дискомфорт у ділянці живота, раннє насичення їжею, втома, тенденція до появи кровотеч і синців, сповільнення або зупинка росту дитини, болі в кістках, незадовільна успішність у школі. Сімейний анамнез має містити інформацію про наявність рідних братів чи сестер із ХГ, кровне споріднення, етнічну приналежність, близьких родичів із гіперспленією, проведену спленектомію, наявність споріднених із ХГ патологій – ранньої появи симптомів, подібних до хвороби Паркінсона (ХП), хвороби Альцгеймера.
Під час медичного огляду оцінюють антропометричні дані, обстежують абдомінальну ділянку, шкіру (блідість, синці, петехії), опорно-руховий апарат, нервову систему, серце та легені. Пальпацію живота потрібно починати з пахових ділянок, щоб не пропустити край значно збільшеної селезінки і печінки. Відсутність неврологічних симптомів у ранньому дитячому віці не виключає наявність неврологічних захворювань, оскільки офтальмологічні симптоми можуть вперше проявитися і в пізньому дитячому віці.
Необхідно виконати аналіз крові, в якому, імовірно, спостерігатиметься анемія, тромбоцитопенія, іноді лейкопенія. Також може бути нехарактерний дефіцит фактора ХІ, який часто спостерігають в ашкеназьких євреїв.
Остаточний діагноз встановлюють після визначення рівня активності кислої b-глюкозидази (глюкоцереброзидази) у лейкоцитах, мононуклеарах, фібробластах і/або проведення молекулярного генетичного аналізу. Аспірація і/або біопсія кісткового мозку є інвазивними маніпуляціями і не рекомендуються для виконання з діагностичною метою [12]. Пацієнтам із типом ІІІ ХГ для ведення моніторингу прогресування патології треба обов’язково визначити підтип захворювання.
Коментар робочої групи. Встановлення діагнозу ХГ у дорослих ґрунтується на тих самих лабораторних дослідженнях, що і в дітей. На момент розробки цієї клінічної настанови в Україні єдиною установою, де проводять ферментодіагностику ХГ, є Центр орфанних захворювань НДСЛ «ОХМАТДИТ» (м. Київ). Тут визначають активність:
- глюкоцереброзидази в лейкоцитах крові або фібробластах шкіри (при ХГ результат дослідження становить <30% від нормальної активності ферменту);
- хітотріозидази (збільшення ферментативної активності >200 Нмоль/год/мл плазми).
За міжнародним класифікатором рідкісних захворювань ORPHANET ХГ має шифр ORPHA:355.
Зростає кількість дітей, в яких діагностують ХГ в досимптомному періоді (за допомогою ферментного чи молекулярного аналізу) шляхом генетичного скринінгу батьків, пренатальної/неонатальної діагностики в спільнотах євреїв ашкеназі або за наявності хворих родичів. Скринінг лізосомальних порушень у новонароджених дає можливість виявити й інших дітей із безсимптомним перебігом захворювання.
Коментар робочої групи. В Україні скринінг на ХГ у новонароджених не проводиться. Ферментний і/або молекулярний аналіз ХГ у досимптоматичному періоді в Україні здійснюється лише в Центрі орфанних захворювань НДСЛ «ОХМАТДИТ».
Молекулярний аналіз і генетичне консультування
Молекулярний аналіз гена кислої β-глюкозидази (глюкоцереброзидази, GBA) рекомендовано проводити кожній дитині з підтвердженим діагнозом ХГ (у тому числі після обстеження хворих без симптомів за допомогою ферментного аналізу), щоб допомогти відрізнити ненейропатичну форму захворювання (тип І) від хронічної нейропатичної (тип ІІІ), які в ранньому дитинстві можуть мати однаковий клінічний перебіг. Хоча генотипово-фенотипові кореляції не є абсолютними, принаймні одна мутація N370S захищає від розвитку нейропатичного типу хвороби, тоді як щонайменше одна мутація L444P має тенденцію до посилення тяжкості патології. Більшість пацієнтів, які були виявлені під час скринінгу спільноти євреїв ашкеназі, мали мутації N370S. Нейропатичний фенотип зазвичай пов’язаний із гомозиготною або компаундною гетерозиготною мутацією L444P і D409H; дані стосовно бразильських пацієнтів також свідчать, що нейропатичну форму ХГ здатна спричинити й мутація G377S. Знання генотипу хворих може бути корисним під час проведення сімейного скринінгу та пренатальної діагностики.
Батькам дитини потрібно запропонувати генетичне консультування лікаря, що має досвід ведення пацієнтів із ХГ. Якщо діагноз був встановлений шляхом скринінгу певної спільноти, генетичне консультування необхідно виконувати до настання вагітності. Пари, де обидва партнери є носіями мутації, мусять знати, що кожна їх спільна дитина матиме ризик успадкування ХГ на рівні 25%. З парами, які є носіями мутації N370S, надзвичайно важливо проводити консультації стосовно широкого спектра симптомів захворювання та визначення віку їх першого прояву.
The Belgian Working Group on Gaucher Disease, Guidelines for diagnosis, teatment and monitoring of Gaucher’s disease (2016)
Генетичні особливості:
- Ген глюкоцереброзидази (GBA1) розташований на хромосомі 1q21.
- Патогенні варіанти N370S, L444P, 84GG, IVS2+1G>A являють собою, відповідно, 90% мутантних алелів в євреїв ашкеназі та 50-60% мутантних алелів в осіб із ХГ типу І, які не є євреями. Однак негативний результат скринінгу на ці поширені мутації не виключає ХГ. Тому в разі серйозної підозри рекомендується секвенування всього гена GBA.
- Поширеність носійства ХГ в певних популяціях (наприклад, 1/18 в євреїв ашкеназі) є високою [5].
- Мутації зумовлюють розподіл ХГ на 3 класи: легкі, середньотяжкі і тяжкі (нульові). Тяжкість певного генотипу важко передбачити, а рідкісність деяких мутацій робить кореляцію генотип-фенотип суперечливою.
- При ненейронопатичній ХГ І типу: мутація N370S була визнана легшою. У пацієнтів щонайменше з одним алелем N370S неврологічні симптоми не розвиваються. У пацієнтів, гомозиготних за мутацією N370S, клінічний перебіг легший, ніж у гетерозигот за цією мутацією. Були також описані пацієнти з генотипом N370S/N370S, в яких не виникало жодних симптомів.
- У популяції осіб, які не є євреями, поширеність алеля N370S є вищою серед португальців та іспанців.
- При нейропатичній формі ХГ ІІІ типу: мутація c.1448T>C (L444P) найчастіше спостерігається у шведів. Пацієнти з щонайменше однією патогенною мутацією L444P мають вищий ризику розвитку неврологічних порушень. Гомозиготність за L444P – це генотип, який найчастіше асоціюється з ХГ ІІ типу і являє собою тяжку патологію з імовірністю неврологічного порушення в дуже молодому віці. Рідкісні генотипи, такі як гомозиготність за D409H та D409H/L444P, також асоціювались із цим фенотипом.
- Гомозиготність за D409H спостерігається рідко та являє собою підтип ХГ ІІІ типу [10]. Для цього фенотипу характерні кальцифікація серцевих клапанів, ішемічна хвороба серця, помутніння рогівки та супрануклеарна офтальмоплегія.
- Припущення про тяжкість нульової мутації, наприклад мутації зі зсувом рамки зчитування або рекомбінантних алелів 84GG, ґрунтується на тому факті, що в гомозиготних пацієнтів із нульовою мутацією зазвичай розвивається тяжкий фенотип і вони помирають під час пренатального періоду або на початку життя, оскільки гомозиготність, імовірно, не сумісна з виживанням/життям.
- Кореляція генотип/фенотип є не зовсім точною, але характеристика генотипу має певну прогностичну цінність.
- Клінічна варіабельність проявів хвороби у братів і сестер відображає неповне розуміння проблеми і може бути пояснена генетичними модифікаторами, однак ця теорія все ще потребує підтвердження.
- Хвороба Паркінсона: більш поширена в гетерозиготних носіїв мутації гена GBA порівняно із загальною популяцією. Мутації гена GBA, окрім N370S, асоціюються з підвищеним ризиком розвитку ХП.
- У деяких пацієнтів із дефіцитом сапозину С спостерігали атипову форму ХГ з нормальною активністю глюкоцереброзидази. У декого з пацієнтів із дефіцитом активатора ферментів були виявлені складні гетерозиготні мутації гена PSAP.
Коментар робочої групи. Зазначають, що 40,4% (61/124) мутантних алелів гена GBA у пацієнтів із ХГ в Україні мають рідкісні перебудови, які спостерігалися в поодиноких випадках. Сумарна частота мажорних мутацій р.N409S та L483P гена GBA в Україні нижча за середньоєвропейську і становить 50,8% (63/124) від усіх ідентифікованих алелів. 12,9% варіантів гена GBA у пацієнтів із ХГ в Україні становили рекомбінантні алелі, які утворились унаслідок конверсії послідовності функціонального гена і високогомологічного псевдогена GBAP. Виявлена висока, порівняно з даними інших європейських дослідників, частота місенс-замін р.R159W та p.G416S (по 4,8%).
Revised recommendations for the management of Gaucher disease in children. European Journal of Pediatrics (2013)
Початкове обстеження хворих із підтвердженим діагнозом
Ретельна первинна оцінка стану здоров’я має важливе значення для всіх дітей зі встановленим діагнозом ХГ, навіть якщо патологія перебуває в досимптомному періоді (Панель 1). Обстеження передбачає аналізи крові, визначення об’єму селезінки та печінки, оцінку захворювань кісток.

Аналіз крові
Лабораторні дослідження мають охоплювати розгорнутий аналіз крові, вивчення функції печінки та нирок, визначення рівнів інших біомаркерів. Серед можливих відхилень може бути високий рівень імуноглобулінів, низький рівень холестерину, особливо ЛПВЩ, збільшений протромбіновий час (у 30% пацієнтів), підвищений рівень біомаркерів (хітотріозидази, CCL18, тартрат-резистентної кислої фосфатази [ТРКФ] та ангіотензин-перетворювального ферменту [АПФ]). У приблизно 6% осіб не експресується хітотріозидаза (фермент, що його секретують активовані макрофаги, який гідролізує трисахариди, отримані з хітину) через наявність нульової мутації гена; у деяких країнах, наприклад на Тайвані, подібне явище спостерігають приблизно в 30% популяції. Для таких пацієнтів із ХГ дослідження біомаркерів АПФ і ТРКФ доцільно виконувати під час проведення безперервного моніторингу проявів захворювання та вивчення ефективності терапевтичного втручання.
Коментар робочої групи. В Україні з переліку рекомендованих біомаркерів визначають лише рівень хітотріозидази в плазмі крові. Обстеження проводять у лабораторії Центру орфанних захворювань НДСЛ «ОХМАТДИТ». Там же проводиться ідентифікація мутації 24bp гена хітотріозидази, яка призводить до відсутності хітотріозидазної активності в плазмі крові, що може спричинити хибно негативну діагностику ХГ. Проведені дослідження показали, що орієнтовна частота нульової мутації гена хітотріозидази в населення України становить 10,1%. Це доводить необхідність проведення молекулярного дослідження на наявність цієї мутації.
Перед початком лікування рекомендується відібрати початковий зразок сироватки (3 мл), який у подальшому слугуватиме контрольним стандартом, та зберігати його в банку крові за температури -70 °C. У разі появи в майбутньому побічних реакцій після ФЗТ чи проведення інших аналізів/досліджень дуже важливе значення має можливість визначення початкового рівня антитіл у сироватці хворого.
Обстеження селезінки, печінки та кісток
За допомогою МРТ необхідно дослідити об’єм та інфільтрацію селезінки і печінки. За відсутності МРТ певне уявлення про розмір органів може дати ультразвукове дослідження (УЗД) (менш точний метод).
Обстеження кісток в ідеалі має передбачати визначення щільності кісткової тканини, виконаної разом із двохенергетичною рентгенівською денситометрією (двохенергетичною рентгенівською абсорбціометрією – ДЕРА) усього тіла, поперекового відділу хребта і/або стегна, МРТ поперекового відділу хребта і стегнових кісток.
Коментар робочої групи. Через недостатність ресурсного забезпечення двохенергетична рентгенівська денситометрія в Україні виконується обмежено.
The Belgian Working Group on Gaucher Disease, Guidelines for diagnosis, treatment and monitoring of Gaucher’s disease (2016)

Коментар робочої групи. Метод СКК не підтверджує діагноз ХГ і потребує подальшого лабораторного обстеження з ретельним зіставленням клінічних симптомів і результатів лабораторних досліджень.
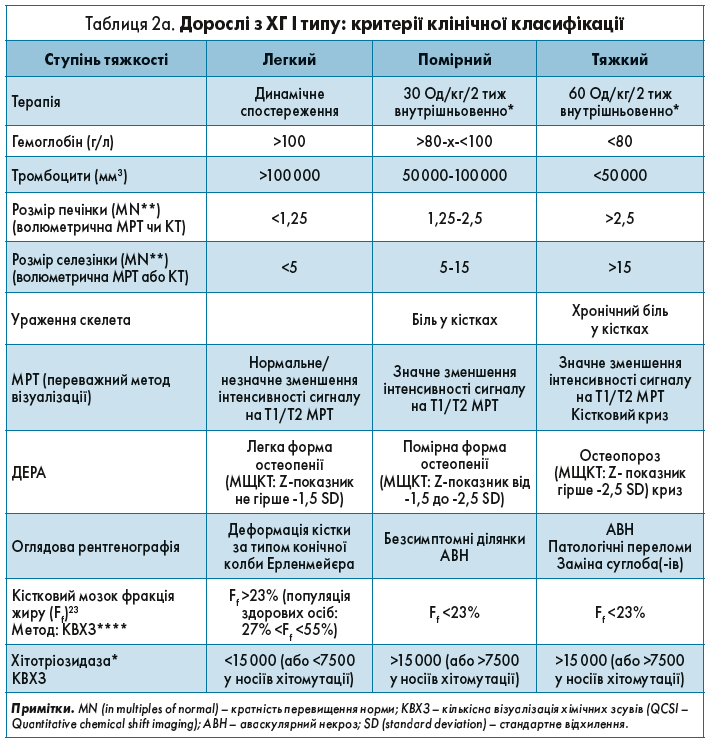
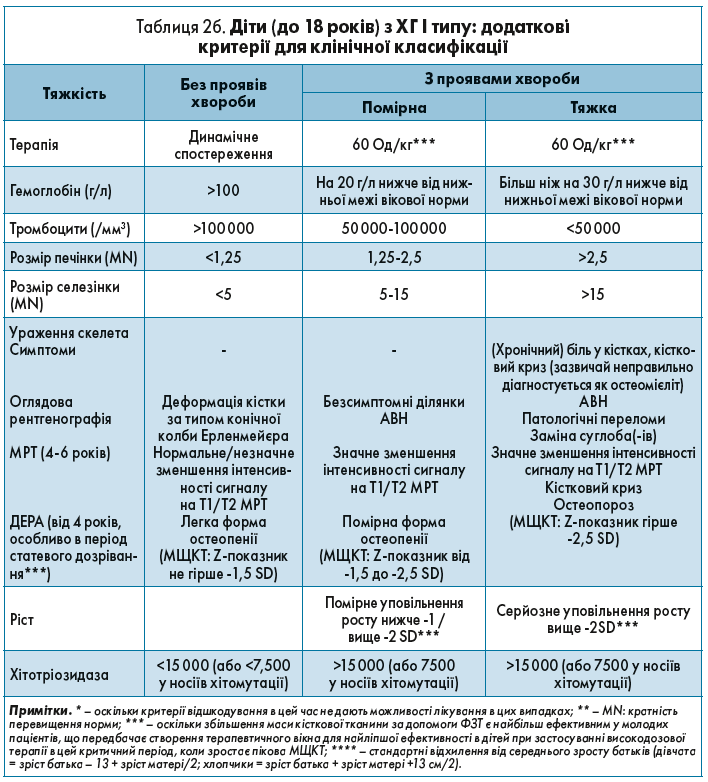
Критерії клінічної класифікації
Рекомендації щодо лікування пацієнтів із ХГ
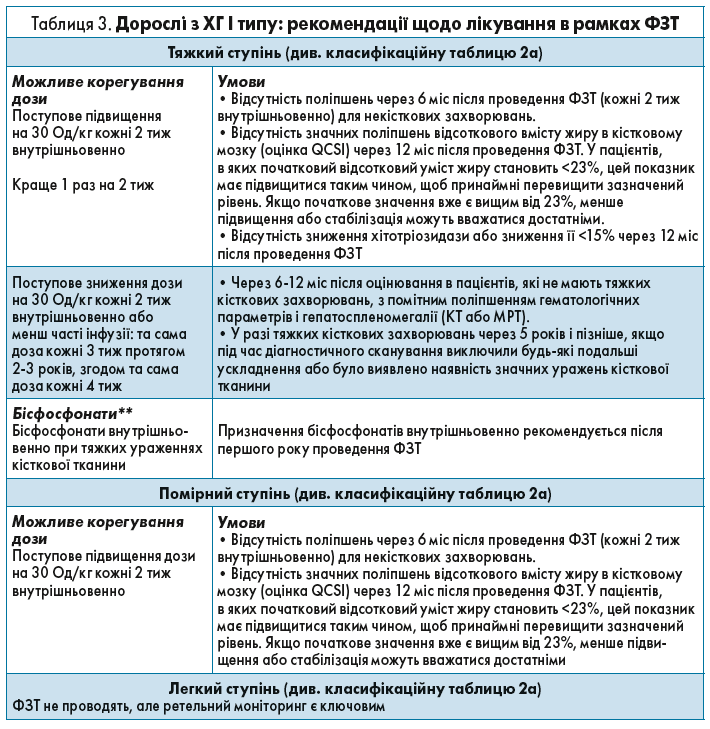
Коментар робочої групи. З метою поліпшення якості життя пацієнтів із ХГ можна змінити кратність інфузії із застосуванням сумарної дози препарату, після стабілізації основних клінічних і лабораторних показників.
Рекомендовані в настанові Belgian expert opinion for diagnosis, treatment and monitoring of Gaucher’s disease 2016 року біcфосфонати в лікуванні тяжкої форми ХГ в Україні не призначалися. Робоча група рекомендує індивідуальний підхід у застосуванні бісфосфонатів при тяжкому перебігу ХГ з ураженням кісткової тканини не раніше ніж через рік після початку ФЗТ. В Україні off lable бісфосфонати застосовували при недосконалому остеогенезі (http://www.mif-ua.com/archive/article/34331).
Коментар робочої групи. Клінічних досліджень для порівняння ефективності трансплантації стовбурових клітин (ТСК) з іншими методами (ФЗТ чи субстрат-редукуюча терапія [СРТ]) не проводилось. ТСК використовувалась як заміна загальноприйнятим методам лікування у тяжких випадках за відсутності доступу чи неефективності лікування. Вона добре показала себе при лікуванні нейропатичних форм ХГ стабілізацією неврологічних проявів. Але використання ТСК обмежене через тяжкість і складність проведення процедури, смертність, ускладнення та відсутність доступних донорів.
Revised recommendations for the management of Gaucher disease in children. European Journal of Pediatrics (2013)
Ферментна замісна терапія
Результатом ФЗТ є розщеплення накопиченого глюкозилцераміду та поліпшення або нормалізація вісцеральних, кісткових симптомів і зростання дитини. У більшості літературних джерел описаний ефект ФЗТ з використанням алглюцерази (Цередаза (Ceredase®), Genzyme Corporation) – глюкоцереброзидази, отриманої з людської плаценти, що впливає на макрофаги і стала доступною в 1991 р., – та її рекомбінантного наступника, іміглюцерази (Церезим (Cerezyme®), Genzyme Corporation), що її застосовують із 1994 року. Альтернативна форма рекомбінантної людської глюкоцереброзидази – велаглюцераза альфа (ВПРІВ (VPRIV®), Shireplc) доступна з 2010 року. Третя рекомбінантна форма людської глюкоцереброзидази – таліглюцераза альфа (Pfizer/Protalix BioTherapeutics, Inc.) отримала схвалення FDA у травні 2012 року.
Кожній дитині та підлітку з наявністю симптомів ХГ треба призначати регулярні внутрішньовенні інфузії ФЗТ. Показання до початку ФЗТ в дітей із наявністю симптомів наведено на Панелі 2.

Дозу підбирають індивідуально відповідно до клінічного статусу пацієнта та його молекулярних показників. Рекомбінантні ферменти доступні більш ніж у 80 країнах світу, у тому числі в Європі. Оскільки лікування є дуже дорогим, процедури відшкодування витрат, схеми і дози початкового лікування різні в різних регіонах. Під час кожного медичного огляду вимірюють вагу дитини та призначають відповідну кількість одиниць препарату на кілограм маси тіла. Постійна катетеризація вени не потрібна, винятками є випадки ускладненого доступу до вени чи виражений страх дитини перед ін’єкцією навіть після попереднього оброблення шкіри місцевим анестетиком, наприклад лідокаїном.
Типовий результат ФЗТ в дітей демонструє суттєве зменшення розмірів селезінки і печінки, а також регресію анемії та тромбоцитопенії. Дані довготривалого спостереження за 884 дітьми, залученими до Реєстру пацієнтів із ХГ (ICGG Gaucher Registry) і які отримували алглюцеразу/іміглюцеразу, показали, що більшість позитивних змін гематологічних показників і приблизно половина поліпшень органомегалії зафіксовані протягом першого року лікування, при цьому параметри продовжували поліпшуватись або залишалися на тому самому рівні протягом щонайменше 8 років терапії (рис. 2 і 3).
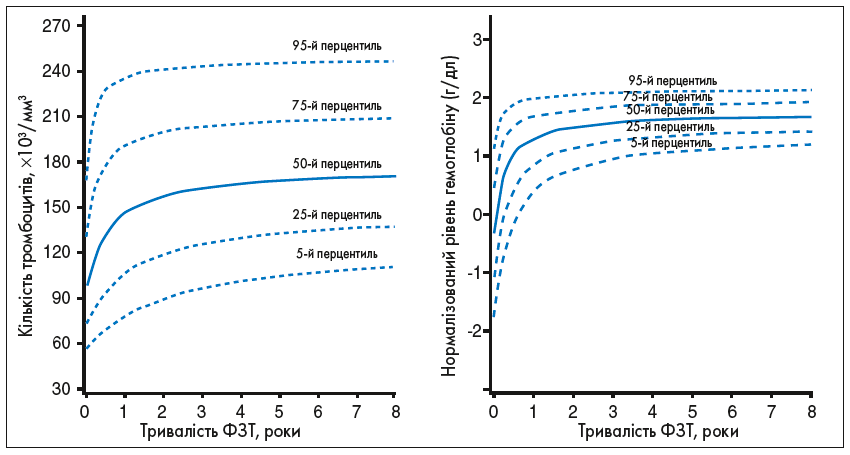
Рис. 2. Зміни рівнів тромбоцитів та гемоглобіну залежно від часу (у роках) лікування іміглюцеразою/алглюцеразою. Відтворено з дозволу Андерсон та співавт. (Andersson et al.) [1].
Дані 884 дітей, що залучені до Реєстру пацієнтів із ХГ, станом на 6 січня 2006 року, які мали інтактну селезінку та отримували іміглюцеразу. Дані щодо вмісту тромбоцитів доступні від 768 пацієнтів (7991 спостереження); дані нормалізованого рівня гемоглобіну доступні від 771 хворого (8022 спостереження). Рівні нормалізованого гемоглобіну проаналізовані в г/дл нижче нижньої межі контрольного діапазону. При цьому керувалися такими значеннями норми, скорегованими за віком та статтю: від народження до 6 міс: <10,1 г/дл; від 6 міс до 2 років: <9,5 г/дл; >2-12 років: <10,5 г/дл; 12 років, хлопчики: <12 г/дл; >12 років, дівчата: <11 г/дл.
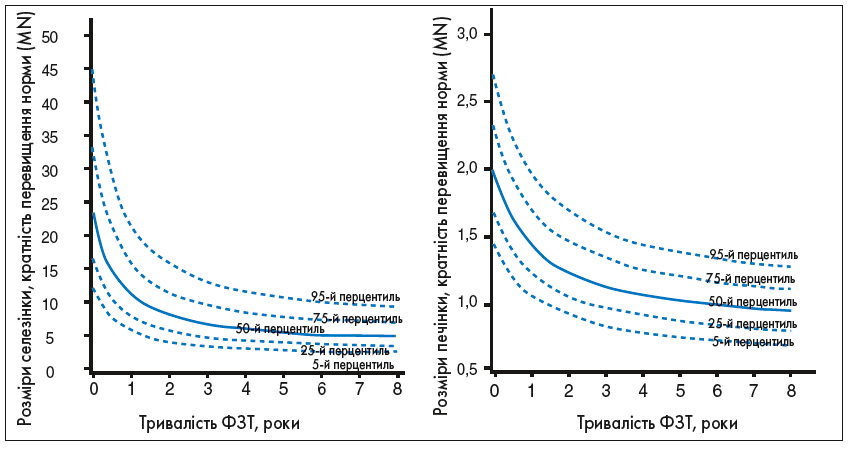
Рис. 3. Зміни розмірів печінки та селезінки залежно від часу (у роках) лікування іміглюцеразою/алглюцеразою. Відтворено з дозволу Андерсон та співавт. (Andersson et al., 2013) [1].
Дані 884 дітей, що залучені до Реєстру пацієнтів із ХГ, станом на 6 січня 2006 року, які мали інтактну селезінку та отримували іміглюцеразу. Дані щодо розміру селезінки доступні від 458 пацієнтів (1593 спостереження); дані щодо розміру печінки – від 420 пацієнтів (1524 спостереження).
Revised recommendations for the management of Gaucher disease in children. European Journal of Pediatrics (2013)
Цей аналіз також продемонстрував поліпшення середніх Z-показників зросту на 1,9 одиниці протягом 8 років лікування алглюцеразою/іміглюцеразою (рис. 4).
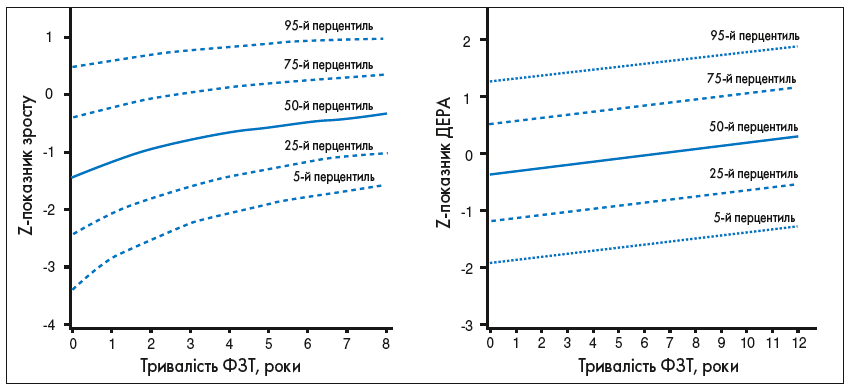
Рис. 4. Зміни Z-показників зросту та МЩКТ хворих залежно від часу (у роках) лікування іміглюцеразою/алглюцеразою. Відтворено з дозволу Андерсон та співавт. (Andersson et al.) [1].
Дані 884 дітей, що залучені до Реєстру пацієнтів із ХГ, станом на 6 січня 2006 року, які мали інтактну селезінку та отримували іміглюцеразу. Дані Z-показників зросту доступні від 702 пацієнтів (5602 спостереження); дані Z-показників МЩКТ – від 127 пацієнтів (244 спостереження).
Графіки на рисунках 2-4 можна використовувати для оцінки індивідуальної відповіді хворого на терапію за тією самою методикою, що й діаграми росту.
У дітей із ХГ, що почали отримувати терапію іміглюцеразою в перші 10 років життя, спостерігається нормальне статеве дозрівання, тоді як у хворих, які не отримували лікування до настання підліткового віку, статеве дозрівання затримується. Оскільки більшість кісткової маси формується протягом пубертатного періоду і досягає максимуму в 30 років, у пацієнтів із ХГ за відсутності лікування неможливість накопичення максимуму кісткової тканини в критичний віковий період разом з остеонекрозом підвищує ризик переломів, у тому числі мікротріщин. ФЗТ збільшує МЩКТ, кісткову масу та щільність кортикального шару, при цьому найбільш виражене поліпшення МЩКТ спостерігали в пацієнтів молодшого віку, що отримували іміглюцеразу. Іміглюцераза також усуває найгостріші кісткові кризи і запобігає появі болю в кістках і переломів. Таким чином, шляхом поліпшення МЩКТ та зменшення кісткових кризів ФЗТ може запобігти виникненню таких серйозних скелетних ускладнень, як компресія суглоба чи хребців та переломів.
У результаті ФЗТ спостерігається повернення до майже нормальних значень підвищених показників таких біомаркерів захворювання, як хітотріозидаза, АПФ, ТРКФ та CCL18. Також у результаті лікування виявляється зниження гіперімуноглобулінемії та високого рівня феритину. Постійна терапія з використанням іміглюцерази поліпшує або нормалізує рівні IgA та IgM, тоді як IgG є менш сприйнятливим до лікування.
Приблизно в 7% пацієнтів спостерігали періодичні побічні реакції під час або через декілька днів після проведення інфузії препарату. Подібним несприятливим реакціям гіперчутливості або явищам неалергічного походження можна запобігти шляхом повільного введення розчину і збільшення часу інфузії і/або попереднього введення антигістамінних препаратів чи кортикостероїдів. Спираючися на багаторічний досвід застосування іміглюцерази, встановлено, що схема введення препарату, яка передбачає початкову повільну швидкість введення з поступовим її підвищенням, є профілактикою виникнення побічних реакцій. Як правило, обстеження та первинне лікування дітей із ХГ проводять в умовах стаціонару, де виконується ретельний моніторинг стану хворих. За відсутності побічних ефектів ФЗТ через 6-12 міс у деяких країнах хворим дозволяється проводити подальше лікування вдома. Такий підхід допомагає поліпшити дотримання лікувального режиму та якість життя пацієнтів і їхніх родин. Тривалість госпітальних інфузій необхідно скорегувати в індивідуальному порядку для кожного хворого до переведення його на домашній режим лікування. Якщо протягом 6-12 міс не виникали побічні реакції, пов’язані з введенням препарату, поява в подальшому антитіл класу IgE малоймовірна.
Продовження у наступному номері.
Текст адаптовано та уніфіковано відповідно до стандартів Тематичного випуску «Медичної газети «Здоров’я України».
Повний текст настанови за посиланням https://www.dec.gov.ua/wp-content/uploads/2023/02/2022_12_06_kn_hvoroba-goshe.pdf
Тематичний номер «Діабетологія. Тиреоїдологія. Метаболічні розлади» № 2 (66) 2024 р.