


19 травня, 2021
Ендотеліальна дисфункція: головний фактор SARS-CoV‑2 (COVID‑19)?
Коронавірусна хвороба 2019 (COVID‑19) спричинила кризу громадського здоров’я пандемічного масштабу. Найчастішими симптомами інфекції, спричиненої коронавірусом 2, тяжкого гострого респіраторного синдрому (SARS-CoV‑2) є кашель, лихоманка та задишка, але можуть також спостерігатися позалегеневі симптоми, зокрема неврологічні та гастроентерологічні прояви. Патогенез захворювання переважно залежить від механізмів надходження та дії коронавірусів. До 2003 року були відомі два типи поверхневих рецепторів коронавірусу. Низці коронавірусів I групи, наприклад коронавірусу людини 229E та вірусам, які спричинюють трансмісивний гастроентерит і котячий інфекційний перитоніт, для проникнення в клітини-мішені потрібна металопротеаза цинку амінопептидаза N (APN, CD13) [1, 2]. Вірус II групи, коронавірус мишачого гепатиту (MHV), використовує представників надродини рецепторів імуноглобулінів, таких як молекули адгезії клітин, пов’язаних із мишачим карциноембріональним антигеном (CEACAM) [3]. Lі та співавт. [4] було ідентифіковано окремий коронавірус як етіологічний агент SARS 2003 року, SARS-CoV‑1, який використовує поверхневий глікопротеїн, так званий spike (S), для доступу до клітин-хазяїнів.
Ключові слова: коронавірусна хвороба, COVID‑19, синдром коронавірусу, SARS-CoV, дисфункція ендотелію, легеневі судинні зміни
Цікаво, що SARS-CoV‑2 тісно пов’язаний із SARS-CoV‑1, і було продемонстровано, що зв’язування S-білків коронавірусів із клітинними рецепторами опосередковує інфікування клітин-мішеней [5]. Lі та співавт. змогли показати, що металопептидаза (ангіотензин-перетворювальний фермент 2, АПФ‑2) [6], виділена з пермісивних щодо SARS-CoV клітин Vero E6 (клітинна лінія клітин нирок африканської зеленої мавпи), може ефективно зв’язувати домен S1 білка SARS-CoV S і діяти як функціональний ко-рецептор для проникнення коронавірусу. Нещодавно було продемонстровано, що АПФ‑2 є геном, стимульованим людським інтерфероном, і це свідчить про те, що SARS-CoV‑2 під час ураження легень може використовувати для посилення інфекції видоспецифічну стимуляцію інтерфероном експресії АПФ‑2, тканинно-захисного медіатора [7].
Патогенез захворювання також залежить від локалізації ко-рецепторів коронавірусу. Як показали Hamming та співавт. [8], АПФ‑2 у людини у великій кількості наявний в епітеліальних клітинах легень і тонкої кишки, тобто клітинах, які контактують із зовнішнім середовищем і можуть стати шляхами проникнення для SARS-CoV‑2. Ця епітеліальна експресія забезпечує перший крок у розумінні патогенезу основних проявів SARS, зокрема в легенях (кашель, пневмонія та тяжкий гострий респіраторний синдром). Пневмоцити I та II типу експресують АПФ‑2 у значній кількості, що вказує на те, що альвеолярні пневмоцити є можливим місцем входу для SARS-CoV‑2. Попадання вірусу може спричинювати цитопатологічні зміни на епітеліальній альвеоло-капілярній поверхні, що спочатку призводить до індукції альвеолярних клітин II типу як першої спроби відновлення. У разі SARS значуща експресія АПФ‑2 в альвеолярних клітинах II типу може спровокувати швидке поширення вірусу та локальне руйнування стінки альвеоли; це призводить до швидко прогресуючого тяжкого дифузного пошкодження альвеол і гіперзапалення, відомого як синдром цитокінової бурі [9]. Ба більше, як було продемонстровано, оксидативний стрес, індукований SARS-CoV‑2, може посилити дефект метилювання ДНК, а це, своєю чергою, може спричинити деметилювання АПФ‑2 та посилення віремії [10]. Оксидативний стрес у легенях виникає, коли антиоксидантні можливості перевантажені або виснажені через зовнішні впливи, як-от зміна парціального тиску кисню або забруднене повітря, чи внаслідок внутрішньої активації резидентних або запальних клітин, залучених у відповідь на вплив чинників навколишнього середовища, травму чи інфекцію [11, 12]. Abouhashem та співавт. [13] нещодавно показали за допомогою даних РНК-секвенування окремих клітин легень людини, що специфічні компоненти антиоксидантної захисної системи альвеолярних клітин II типу, а саме супероксиддисмутаза 3 та активувальний фактор транскрипції 4, сенсор стресу ендоплазматичного ретикулума, слабшають у відповідь на старіння в пацієнтів похилого віку. Це може частково впливати на тяжкість перебігу COVID‑19 у літніх людей.
Мішенями SARS-CoV є також імунні органи та системні дрібні судини, що призводить до системного васкуліту та зниження імунної функції. Інші рецептори/посередники на поверхні клітин людини, імовірно, сприяють надходженню SARS-CoV‑2, у тому числі трансмембранна серинова протеаза 2 [14], сіалова кислота [15] та індуктор металопротеїнази позаклітинного матриксу (CD147, відомий також як базиджин) [16].
Цікаво, що АПФ‑2, так само як інші три рецептори, наявні на артеріальних і венозних ендотеліальних клітинах та клітинах гладеньких м’язів артерій [8]. АПФ‑2 є найбільш вивченим із цих рецепторів-посередників, і нещодавно були порушені питання щодо використання інгібіторів ренін-ангіотензин-альдостеронової системи, як-от блокатори рецепторів ангіотензину II (БРА) та інгібітори АПФ‑2, у пацієнтів із COVID‑19 і артеріальною гіпертензією [17-23]. Зокрема, дані про потенційний зв’язок між застосуванням інгібіторів АПФ або БРА та ризиком розвитку SARS-CoV‑2 були суперечливими. Нещодавно два окремі дослідження, до яких було залучено велику когорту пацієнтів, продемонстрували відсутність доказів підвищеного ризику розвитку COVID‑19 через призначення інгібіторів АПФ‑2 або БРА [23, 24]. Цікаво, що інтерлейкін‑6 (ІL‑6), який продукується під час синдрому цитокінової бурі, також може бути індукований ангіотензином II внаслідок механізму, що опосередковується рецепторами мінералокортикоїдів [25, 26].
Експресія АПФ‑2 на ендотеліальних клітинах, клітинах гладенької мускулатури та периваскулярних перицитах практично в усіх органах свідчить про те, що SARS-CoV‑2, потрапивши в циркуляцію, може легко поширюватися по організму [8]. Зазначимо, що в нещодавному дослідженні, яке порівнювало зразки тканини легень, відібрані для патологоанатомічного дослідження, пацієнтів, померлих від COVID‑19, гострого респіраторного дистрес-синдрому (ГРДС) внаслідок грипу A (H1N1), та неінфікованих легень відповідних за віком пацієнтів контрольної групи, автори виявили більшу кількість АПФ‑2-позитивних ендотеліальних клітин та суттєві зміни морфології ендотелію з порушенням міжклітинних з’єднань, набряком клітин і втратою контакту з базальною мембраною [27].
Усе більше даних показують, що в пацієнтів із COVID‑19 найпоширенішими супутніми захворюваннями, асоційованими з гіршим прогнозом і вищим рівнем смертності, є системна гіпертензія, діабет та ожиріння, на тлі яких дисфункція ендотелію [28] є ключовим фактором. В Європейському респіраторному журналі (Eur Respir J 2020; 56: 2001634) у двох публікаціях про результати досліджень наголошується про ризик тромбозу глибоких вен та гострої легеневої емболії при COVID‑19 [29, 30]. Ці дані підтверджують і розширюють попередні спостереження стосовно визначальної ролі дисфункції ендотелію, пов’язаної із SARS-CoV‑2, підвищеним ризиком розвитку венозних тромбоемболічних захворювань, системного васкуліту, апоптозу ендотеліальних клітин і запалення в різних органах [31-34]. Відомо також, що SARS-CoV‑2, так само як інші інфекційні збудники, унаслідок запальних реакцій може активувати коагулопатію. Відкриття, що щільні гранули тромбоцитів містять поліфосфати та виділяють їх у разі активації, допомогло виявленню широких зв’язків між імунною системою та коагуляційним каскадом (процесом зсідання крові) [35]. Поліфосфати, вивільняючися з активованих тромбоцитів, прискорюють активацію фактору V, пригнічують антикоагулянтну активність інгібітора тканинного фактору, сприяють активації фактору XI тромбіном і синтезу товстіших ниток фібрину, стійких до фібринолізу [36]. Запальний ефект цитокінів також призводить до активації ендотеліальних клітин судин і пошкодження ендотелію, що зумовлює протромботичний ефект. Пошкодження судинного ендотелію згодом спричинює тромбоцитопенію, зменшення вмісту природних антикоагулянтів, а також гемостатичну активацію як фенотиповий прояв тромботичної дифузної внутрішньосудинної коагуляції. У разі коагулопатії, асоційованої з COVID‑19 [37], механізми, які активують коагуляцію при інфікуванні SARS-CoV‑2, лишаються невідомими, але, схоже, пов’язані, імовірніше, із запальними реакціями, а не зі специфічними властивостями вірусу [38].
Klok та співавт. [39] нещодавно повідомляли про 31% випадків тромботичних ускладнень у пацієнтів із COVID‑19, госпіталізованих у відділення інтенсивної терапії. Grillet та співавт. [40] встановили, що у 23% хворих на COVID‑19-пневмонію була гостра легенева емболія, виявлена за допомогою комп’ютерної томографії та легеневої ангіографії. Крім того, у критичних хворих на COVID‑19 з оклюзією та мікротромбозом дрібних легеневих судин може спостерігатися гострий набряк легень [41]. Інші дослідники висували гіпотезу, що тромби можуть відігравати безпосередню й значну роль у порушеннях газообміну та мультисистемних дисфункціях органів у разі COVID‑19-пневмонії [42]. Ба більше, декілька авторів висловлювали припущення про роль антифосфоліпідних антитіл у тромботичних подіях, пов’язаних із COVID‑19 [43-45]. Поки що залишається дискусійним питання, чи доцільно вважати наявність цих антитіл доказом ранньої антикоагулянтної активності в пацієнтів із COVID‑19, знаючи, що антифосфоліпідні антитіла поширені в загальній популяції, особливо під час інфекції [46].
Gattinoni та співавт. [47] наголосили, що в пацієнтів із COVID‑19-ГРДС спостерігають розбіжність між відносно добре збереженою механікою легень і тяжкою гіпоксемією. Ці автори припустили, що така тяжка гіпоксемія, що виникає в піддатливих легенях, може бути наслідком втрати регуляції перфузії легень і гіпоксичного звуження судин. Це нагадує ослаблену гіпоксичну легеневу вазоконстрикцію внаслідок дисфункції легеневих клітин ендотелію, спричиненої підвищеним окислювальним стресом мітохондрій у гризунів, що зазнали дії дазатинібу [48]. Нарешті, було показано, що елементи SARS-CoV‑2 можуть бути виявлені в ендотеліальних клітинах пацієнтів із COVID‑19 разом із накопиченням запальних клітин та свідченням загибелі ендотеліальних та запальних клітин [49]. Це свідчить про те, що ендотеліїт може спричинюватися SARS-CoV‑2 та розвиватися в різних органах як прямий наслідок вірусної інфекції та запальної реакції хазяїна. Згідно з цим Ackermann та співавт. [27] виявили, що крім дифузного пошкодження альвеол із периваскулярною Т-клітинною інфільтрацією та поширеного тромбозу з мікроангіопатією характерні особливості судин у пацієнтів із COVID‑19 полягали в тяжкому ураженні ендотелію, зумовленому наявністю вірусу в клітинах і пошкодженням клітинних мембран, а також в ангіогенезі.
Зрештою, наявність SARS-CoV‑2 в ендотеліальних клітинах свідчить про те, що прямі вірусні ефекти, так само як периваскулярне запалення, можуть призвести до пошкодження ендотеліальних клітин. Цілком імовірно, що ендотеліїт, ушкодження ендотелію, дисфункція ендотеліальних клітин і порушення мікроциркуляції в різних судинних руслах грають неабияку роль у розвитку загрозливих для життя ускладнень COVID‑19, зокрема венозної тромбоемболії та поліорганної недостатності (рис.).
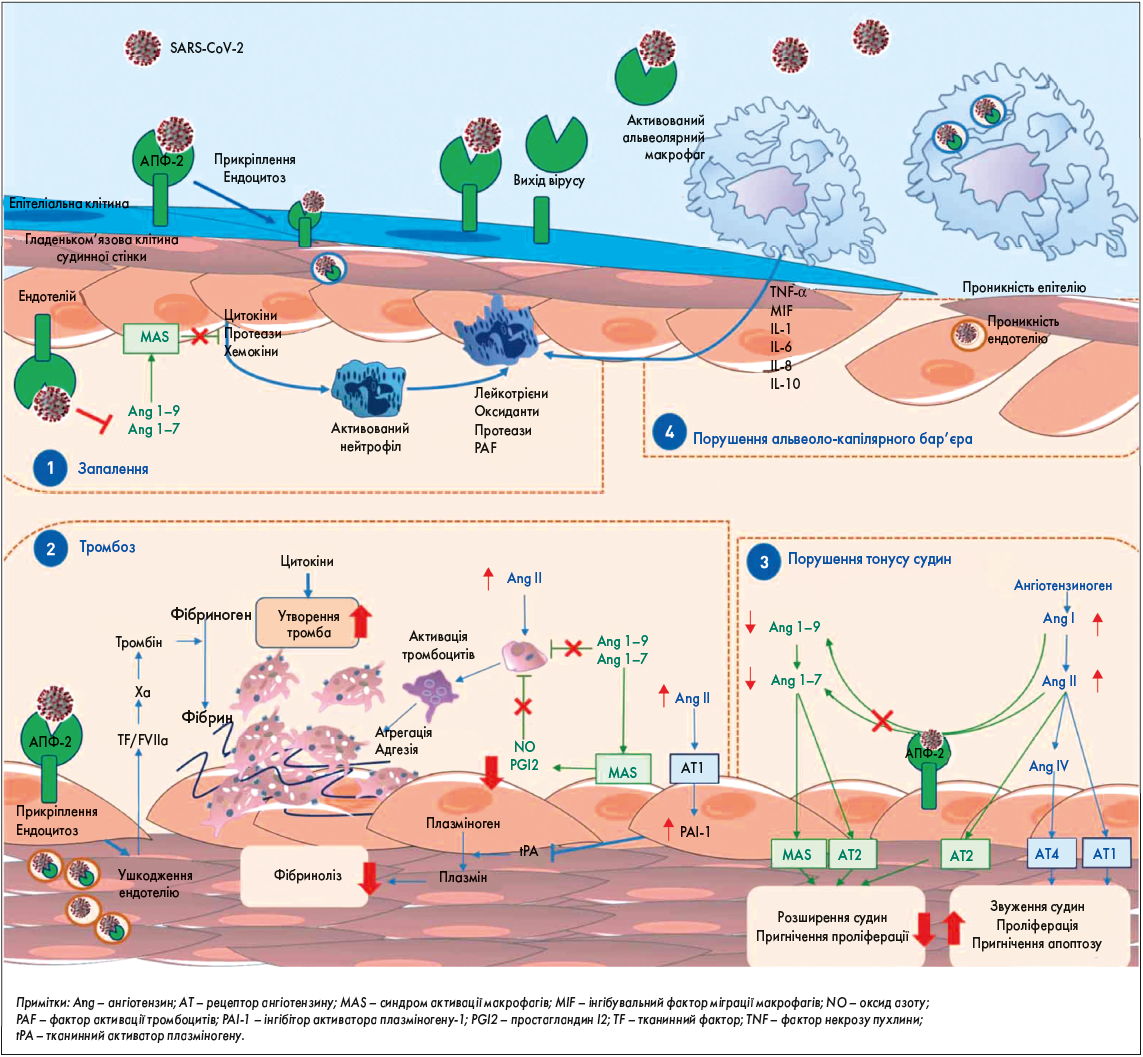
Рис. Схематичне зображення гіпотетичних механізмів, за допомогою яких SARS-CoV‑2 спричинює дисфункцію ендотелію та зміни в судинах легень
Як повідомлялось, після розщеплення S-білка SARS-CoV‑2 потрапляє в клітини людини через зв’язування з АПФ‑2. Цей трансмембранний рецептор широко експресується в різних клітинах легень, у тому числі в альвеолярних клітинах II типу, макрофагах, ендотеліальних, гладеньком’язових клітинах і периваскулярних перицитах. Це спричиняє неконтрольоване запалення (1), яке супроводжується мікротромбозом та оклюзією дрібних легеневих судин (2), а також порушенням ендотеліальної регуляції судинного тонусу (3), що призводить до порушення альвеоло-капілярного бар’єру (4).
Список літератури – у редакції.
Huertas A., Montani D., Savale L. et al. Endothelial cell dysfunction: a major player in SARS-CoV‑2 infection (COVID‑19)? Eur Respir J 2020; 56: 2001634. https://doi.org/10.1183/13993003.01634-2020.
Переклав з англ. Назар Лукавецький