


14 лютого, 2019
Роль ренін-ангіотензин-альдостеронової системи у регуляції артеріального тиску: огляд літератури (продовження)
 В останні роки в процесі дослідження науковці зробили важливе відкриття – проренінові рецептори (ПРР) мають функцію, незалежну від зв’язування реніну та прореніну. Вона полягає в забезпеченні збірки, стабільності та функціонування H+-АТФази вакуолярного типу (V-ATФази) – ферментної системи, що складається з домену протонної транслокації V0, домену помпи V1 та двох допоміжних білків – Ac45 і ПРР.
В останні роки в процесі дослідження науковці зробили важливе відкриття – проренінові рецептори (ПРР) мають функцію, незалежну від зв’язування реніну та прореніну. Вона полягає в забезпеченні збірки, стабільності та функціонування H+-АТФази вакуолярного типу (V-ATФази) – ферментної системи, що складається з домену протонної транслокації V0, домену помпи V1 та двох допоміжних білків – Ac45 і ПРР.
Інакше кажучи, ця система використовує енергію від гідролізу АТФ для отримання протонного градієнта. V-ATФази присутні практично у всіх типах клітин еукаріотів, переважно локалізовані на мембранах внутрішньоклітинних структур і беруть участь у переміщенні везикул, поєднаному транспортуванні та деградації білків [47, 45]. У деяких клітинах вони розміщені в плазматичній мембрані, зокрема в остеокластах, де задіяні в процесах резорбції кісткової тканини, а також у клітинах збірних трубок ниркових канальців, де регулюють кислотно-лужний гомеостаз [49, 50]. Важливість ПРР у функціонуванні цієї системи встановлена після доведення, що його відсутність навіть на тканинному рівні призводить до летальних наслідків. Для прикладу: піддослідні тварини з нокаутом рецептора не виживають на ранніх фазах внутрішньоутробного розвитку, абляція рецепторів у кардіоміоцитах спричиняє смерть від серцевої недостатності (СН), а нокаут у подоцитах – летальні випадки внаслідок гломерулосклерозу в середньому за 3 тижні [51-53, 45].
Ще однією функцією ПРР, незалежною від реніну та прореніну, є участь у сигнальних шляхах Wnt [54]. Білки Wnt мають складну структуру, що нагадує кисть руки. Це фактори росту, необхідні для нормального розвитку ембріона, а у дорослих – для процесів проліферації, міграції та полярності клітин. Порушення передачі сигналів через шлях Wnt спричиняє дегенеративні та онкологічні захворювання у людини. Їхній вплив на клітини-мішені здійснюється через зв’язування з рецепторами – трансмембранним білком Фрайзелда (Fz) та ліпопротеїнами низької густини (LRP 5/6). У шляху Wnt/β-катенін ПРР є адаптером між Fz та LRP 5/6, а протонний градієнт, що генерується V-ATФазою, необхідний для фосфорилювання LRP 5/6 та активації β-катеніну [55].
Ренін належить до родини аспартат протеаз, до якої також відносять пепсин, катепсин та хімозин [33, 34]. Основним компонентом його активного центра є специфічний субпідрозділ (S3sp), відмінний від інших аспартат протеаз, каталітична активність якого зумовлена двома залишками аспарагінової кислоти, що розташовані на кожній із часток. Активний центр може вмістити 7 амінокислотних залишків єдиного відомого на сьогодні субстрату – ангіотензиногену, до якого фермент має високу специфічність. За хімічною структурою ангіотензиноген є глікопротеїном, що містить 452 амінокислотних залишки, належить до α2-глобулінів та синтезується насамперед у печінці, а також у серці, судинах, нирках та жировій тканині. Після розщеплення пептидного зв’язку між лейциновим та валіновим амінокислотними залишками (Лей10-Вал11) в активному центрі реніну ангіотензиноген перетворюється у декапептид ангіотензин І (ангіотензин, ангіотензин1-10) [34, 48].
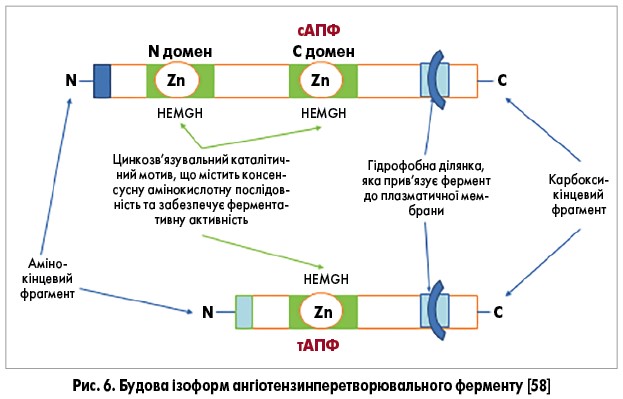
Внаслідок відщеплення С-кінцевого дипептиду від молекули ангіотензин І утворюється октапептид ангіотензин ІІ (ангіотензин1-8). Ця реакція каталізується ангіотензинперетворювальним ферментом (АПФ) з родини цинковмісних металопротеїназ. За хімічною структурою АПФ є дипептидилкарбоксипептидазою, оскільки відщеплює дві амінокислоти з карбоксильного кінця пептидів [56]. Це ектофермент, вбудований у клітинну мембрану, звідки вивільняється у кров. Ідентифіковано дві різні ізоформи АПФ – соматичну (сАПФ) і тестикулярну (тАПФ), які транскрибуються з одного гена на різних сайтах ініціації. Соматична ізоформа – це великий білок з цитоплазматичним хвостом та двома гомологічними каталітичними доменами N і C. Він синтезується в ендотелії судин. Експресію АПФ знайдено у легенях, проксимальних канальцях нирок, серці, дванадцятипалій кишці, запальних клітинах. Порівняно із соматичною ізоформою, тАПФ має у 1,5 раза меншу атомну масу та єдиний каталітичний домен C. Ця ізоформа виявлена лише у дозріваючих і зрілих сперматозоїдах [57] (рис. 6). Раніше каталітичні домени АПФ вважалися еквівалентними, але в останні роки з’ясовано, що ці активні сайти мають цілком різні функції in vivo. Отже, C-кінцевий домен АПФ відповідає за утворення ангіотензину ІІ, а у тестикулярній формі контролює чоловічу фертильність, а саме: здатність сперматозоїдів зв’язуватися з незаплідненими овоцитами [58, 59]. Натомість N-кінцевий домен гідролізує тетрапептид N-ацетил-серил-аспартил-лізил-пролін (AcSDKP), що регулює гемопоетичну проліферацію стовбурових клітин та запобігає проліферації фібробластів у міокарді, аорті, нирці та легенях після їхнього пошкодження [60, 58, 62, 63, 65]. Деградацію брадикініну, субстанції Р, рилізинг фактора лютеїнізуючого гормона можуть каталізувати обидва домени сАПФ [66, 18].
Отже, окрім розщеплення ангіотензин І, соматична форма АПФ гідролізує гептапептид ангіотензин1-9 у біологічно неактивний ангіотензин1-5, інактивує два пептиди-вазодилататори (брадикінін та каллідін), відповідає за деградацію регулятора проліферації стовбурових клітин та фібробластів N-ацетил-серил-аспартил-лізил-проліну, бере участь у біохімічних перетвореннях нейропептидів (нейротензину, енкефалінів), відіграє роль у процесах атерогенезу і запалення [15, 67]. Для прикладу, у макрофагах розірваних і гіперцелюлярних атеросклеротичних бляшок виявлено підвищений вміст АПФ, тоді як у макрофагах фіброзних бляшок кількість АПФ була незначною [68]. Це вказує на роль АПФ у дестабілізації атеросклеротичних бляшок.
Сучасні дослідження продемонстрували, що, окрім гідролітичної функції, АПФ також має здатність передавати внутрішньоклітинні сигнали. Зокрема, зв’язування з ІАПФ (раміприлатом чи периндоприлатом) або брадикініном призводить до фосфорилювання ферменту, що запускає сигнальний каскад в ендотеліальних клітинах. Тобто АПФ є сигнальною молекулою для ІАПФ, що ймовірно забезпечує позитивний вплив цих речовин на серцево-судинну систему, чого не спостерігається при зв’язуванні АПФ з ангіотензином І [69].
Ангіотензин ІІ є потужним вазоконстриктором. Окрім того, через рецептори 1 типу він стимулює виділення клубочковою зоною кори наднирників (zona glomerulosa) гормона альдостерону, сприяючи переміщенню холестеролу у мітохондрії, де останній перетворюється у прегненолон. Внаслідок низки хімічних реакцій утворюється кортикостерон, з якого синтезується альдостерон у присутності альдостерон синтази. Альдостерон індукує експресію трьох генів, пов’язаних з поглинанням води, які забезпечують регуляцію водно-електролітного балансу: ENaC3 – епітеліальний натрієвий канал 3, SGK1 – сироватково / глюкокортикоїд регульована кіназа‑1, натрій-калій АТФаза [70, 71].
Вазоконстрикція та підвищення секреції альдостерону – це швидкі ефекти ангіотензину ІІ. Іншими проявами швидкої дії є: підвищені спрага і потреба в кухонній солі, виділення антидіуретичного гормона, посилення всмоктування натрію в кишечнику, що призводить до затримки рідини; підвищена скоротливість міокарда, що збільшує серцевий викид; підвищення активності симпатичної нервової системи [72]. Тривалими ефектами ангіотензину ІІ є структурні зміни (ремоделювання) серцево-судинної системи: гіперплазія непосмугованих м’язів судинної стінки та міокарда, відкладання компонентів позаклітинного матриксу, сенсибілізація кровоносних судин до низьких концентрацій вазоконстрикторів [15].
Також вченими нещодавно встановлено, що у людини ангіотензин ІІ є потужним конкурентним та селективним інгібітором соматичної ізоформи АПФ з високою селективністю до активного сайту C-домену. Аналіз структури комплексу С-домен АПФ-ангіотензин ІІ показав, що передостанній амінокислотний залишок у молекулі ангіотензину ІІ (пролін) забезпечує стійкість до гідролізу ангіотензину І. Таке пригнічення АПФ ангіотензину ІІ створює механізм негативного зворотного зв’язку, що забезпечує збереження субстрату для альтернативних ферментів (рис. 7) та, очевидно, відіграє роль у природній регуляції гомеостазу компонентів РААС, незалежній від реніну. Оскільки концентрація циркулюючого ангіотензину ІІ досить низька, цей ефект може мати значення у тканинах, зокрема у серці та нирках, де його рівень значно вищий [18].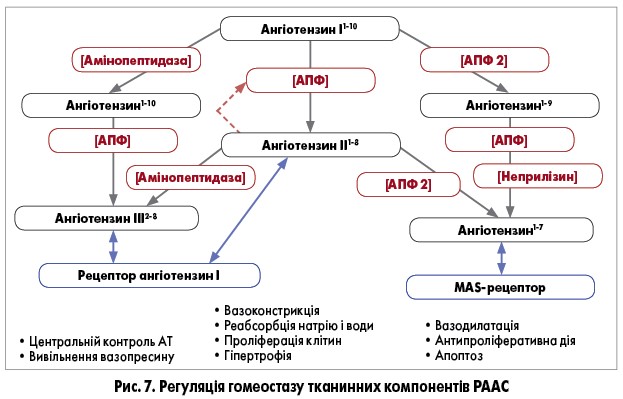
Дія ангіотензину ІІ реалізується через специфічні трансмембранні рецептори чотирьох типів, що розміщені на зовнішніх мембранах ефекторних клітин. Зв’язування ангіотензину ІІ із G-білком рецепторів призводить до їхньої активації. Найбільш вивченими є рецептори першого та другого типів, що виявлені у серці, печінці, нирках, наднирниках, матці, головному мозку, периферійних симпатичних нервах, лімфатичних органах, ендотеліальних і непосмугованих м’язових клітинах судин, фібробластах і макрофагах [73, 74].
Рецептори ангіотензину першого типу (РА1) зумовлюють усі основні ефекти ангіотензину ІІ: вазоконстрикцію, інотропну дію, гіпертрофію міокарда, стимуляцію продукції альдостерону та вазопресину, вивільнення катехоламінів із наднирників і пресинаптичних мембран [75]. Зв’язування ангіотензину II з РА1 спричиняє також прозапальний ефект завдяки активації внутрішньоклітинного сигнального каскаду ядерного фактора транскрипції каппа В (NF-kB), стимуляції секреції фактора некрозу пухлини, інтерлейкіну‑6 та циклооксигенази‑2 та активації системи комплементу [14, 76-78]. Вважається, що ангіотензин ІІ є медіатором дисфункції ендотелію та атерогенезу, оскільки регулює експресію молекул адгезії (VCAM‑1, ICAM‑1, P-селектин), стимулює утворення супероксид-аніонів, перекисне окислення ліпідів та утворення металопротеїназ, інактивує окис азоту (NO) і простациклін, сприяє проникненню моноцитів і лімфоцитів в атеросклеротичну бляшку, підвищує окислення ліпопротеїнів низької густини у макрофагах [79, 80, 81, 82]. Ще однією властивістю ангіотензину II є стимуляція росту клітин. Спричиняючи гіпертрофію мезангiоальних клiтин, він сприяє гломерулосклерозу. Разом з іншими гемодинамічними ефектами РААС це відіграє ключову роль у прогресуванні хронічного захворювання нирок [83]. Блокада РА1 є основним механізмом дії блокаторів рецептора ангіотензину ІІ (БРА ІІ), внаслідок чого відбувається пригнічення вказаних ефектів.
Рецептори ангіотензину ІІ (РА2) вважаються фізіологічною противагою РА1. Активація РА2 може безпосередньо протидіяти РА1-залежним ефектам ангіотензину ІІ через гетеродимеризації обох рецепторів на поверхні клітини. Це спричиняє протизапальну та гіпотензивну дію [84]. Іншим способом антагонізму є блокування сигналів ангіотензину ІІ з РА1 сигналами з РА2 через негативні перехресні перешкоди у цитоплазмі клітин [85]. Найбільше РА2 виявлено у тканинах ембріона, але у перші тижні після народження їхня кількість різко знижується, що свідчить про участь рецепторів у процесах проліферації та диференціації тканин [83, 86]. В організмі дорослої людини РА2 виявлені у мозковому шарі кори наднирників, матці, яєчниках, ендотелії судин, серці та мозку. Їхня активація спричиняє вазодилатацію та гальмування росту клітин [87]. В інтерлобулярних артеріях нирок РА2 пригнічують біосинтез реніну та утворення ангіотензину II, регулюють натрійурез і клубочковий кровотік [88]. Експресія РА2 значно посилюється при інфаркті міокарда, хоча рівень експресії рецепторів цього типу в серці набагато нижчий, ніж у інших тканинах [89, 85]. Точна патофізіологічна роль РА2 вивчена недостатньо, але дослідження in vivo виявили їхню здатність поліпшувати функцію серця після ішемії /некрозу, сповільнювати процеси ремоделювання міокарда і судинної стінки та зменшувати запалення [87].
Рецептори ангіотензину ІІІ виявлені на мембранах нейронів, їхня функція невідома.
Рецептори ангіотензину IV (РА4) – це інсулін-регульована мембранна амінопептидаза, що активується ангіотензином IV. Вони знайдені на ендотеліоцитах, у серці, легенях, нирках, наднирниках та центральній нервовій системі. РА4 беруть участь у регуляції пізнавальних функцій, виділенні окситоцину, експресії молекул адгезії та ймовірно відіграють роль у патогенезі протеїнурії [90-95].
Пептидні ланцюги ангіотензину І та ІІ можуть розщеплюватися у декількох місцях різними видами пептидаз – амінопептидазами, карбоксипептидазами чи ендопептидазами. Утворені пептиди циркулюють у крові та мають ефекти, відмінні від ангіотензину ІІ [96]. Недавно описано нові форми ангіотензину, зокрема продукти розщеплення ангіотензину ІІ, – ангіотензин ІІІ та IV, а також ангіотензин1-5, ангіотензин1-7 та ангіотензин1-9 [97-101].
Ангіотензин III (2-8 пептид) утворюється з ангіотензину ІІ під дією амінопептидази А, яка розщеплює зв’язок аспарагінова кислота1 – аргінін2. Це основний представник РААС у мозку. Порівняно з ангіотензином ІІ, його вазопресивний ефект на 60% слабший, але здатність продукувати альдостерон така сама. Ангіотензин III також стимулює спрагу та виділення вазопресину, не впливає на функцію нирок, але через ниркові РА2 може посилювати натрійурез. Ангіотензин ІІІ та дрібні пептиди, які утворюються внаслідок його метаболізму, здатні зв’язуватися з РА1 та РА2 [96, 102, 718].
Ангіотензин IV (3-8 пептид) є гексапептидом, що утворюється за допомогою відщеплення аргініну від N-кінця ангіотензину ІІІ під дією амінопептидази М або безпосередньо з ангіотензином ІІ під дією D-амінопептидази. Зв’язуючись з РА4, ангіотензин IV активує NF-kB, прозапальні фактори (моноцитарний хемотаксичний білок‑1, інтерлейкін‑6, фактор некрозу пухлини, молекула міжклітинної адгезії‑1) та інгібітор‑1 активатора плазміногена [96]. У серці ангіотензину IV здатний зменшувати викид лівого шлуночка та тиск у ньому, підвищуючи при цьому чутливість до збільшення тиску під час систоли та швидкість релаксації під час діастоли [104]. В артеріях легень цей пептид підвищує активність ендотеліальної NO-синтази та вміст внутрішньоклітинного цГМФ в ендотеліоцитах [105]. У нирках він збільшує кровотік у кірковій речовині, не впливаючи на рівень АТ, і зменшує транспорт Na+ у проксимальних канальцях, але, зв’язуючись з РА1, може спричиняти і вазоконстрикцію [106, 107]. У головному мозку ангіотензин IV бере участь у регуляції локального кровотоку та покращує когнітивні функції при амнезії [108, 109].
Ангіотензин1-7 є активним гептапептидом РААС, що протидіє ефектам ангіотензину II, зокрема спричиняє вазодилатацію. Він може утворюватися двома шляхами: 1) з ангіотензином I за участю нейтральних ендопептидаз, наприклад, неприлізину, ендопептидази 24.11; 2) з ангіотензином ІІ під дією пролілендопептидази, пролілкарбокси-пептидази або АПФ 2, який розщеплює зв’язок пролін7-фенілаланін8 [110, 111, 169]. Іншими властивостями ангіотензину1-7 є кардіоренопротекція, покращення ендотеліальної функції, антигіпертрофічна, антифібротична, антиаритмічна, антитромботична та протизапальна дія, які реалізуються через Mas-рецептори, що включають онкоген-кодований G-білок [113, 114]. Описана також роль цього пептиду при АГ, гіпертрофії міокарда, застійній СН, інфаркті міокарда, хронічних захворюваннях нирок, діабетичній нефропатії, цирозі печінки, гестаційному діабеті та прееклампсії [115-120]. Під дією АПФ ангіотензину1-7 метаболізується у неактивний ангіотензин1-5.
АПФ 2 також каталізує перетворення ангіотензину I в ангіотензин1-9. АПФ 2 має один каталітичний домен, а ген, що кодує її синтез, локалізований у Х-хромосомі. За своєю природою це екзопептидаза, що є інтегральним мембранним глікопротеїном 1 типу та виявляється у більшості тканин з максимальною експресією в ендотелії (особливо в аорті), серці, нирках та гіпоталамусі [99, 121]. У серцево-судинній системі цей фермент, очевидно, має більше значення у регуляції тканинного рівня ангіотензину ІІ, ніж АПФ [122]. АПФ 2 високо специфічний до ангіотензину І та ІІ, але не здатний метаболізувати брадикінін [123]. На ангіотензин1-7 та АПФ 2 не впливають ІАПФ та БРА ІІ. До того ж під впливом цих препаратів рівні АПФ 2 та ангіотензину1-7 підвищуються, особливо у міокарді та нирках [121, 86, 107].
Ангіотензин1-5 та ангіотензин1-9 вважаються неактивними пептидами [15, 61].
Отже, відповідно до сучасних уявлень РААС – це не пресорна система, а система з подвійними функціями. Її судинозвужувальні/проліферативні та судинорозширювальні/антипроліферативні ефекти визначаються балансом між АПФ та АПФ2. Підвищена активність АПФ і/або знижена активність АПФ 2 посилює утворення ангіотензину II та розпад ангіотензину1-7, що спричиняє вазоконстрикцію та стимулює проліферацію, тоді як підвищена активність АПФ2 та/або знижена активність АПФ веде до протилежного ефекту [64, 112].
Список літератури знаходиться в редакції.
Медична газета «Здоров’я України 21 сторіччя» № 21 (442), листопад 2018 р.