


17 січня, 2020
Майстер-клас для лікаря з діагностування хвороби Помпе: аналіз клінічних випадків
Хвороба Помпе (синоніми: генералізований глікогеноз, глікогеноз II типу) – рідкісне нервово‑м’язове захворювання із прогредієнтним перебігом, зумовлене надлишковим накопиченням глікогену лізосомами внаслідок мутації гена GAA і втрати активності ферменту кислої α-глюкозидази. Поширеність цієї патології становить у середньому 1 : 40 тис. осіб (Пічкур Н.О. та співавт., 2017). За даними Державної служби статистики, населення України у 2019 р. нараховувало близько 42 млн. Таким чином, у країні має бути приблизно 1050 пацієнтів із глікогенозом ІІ типу. Але станом на 2017 р. було виявлено лише 5 хворих (Пічкур Н.О. та співавт., 2017). Тож недостатня інформованість населення та медичного персоналу є гострою проблемою в умовах доступного благодійного забезпечення діагностики та лікування хвороби Помпе. Детальний опис клінічних випадків хвороби Помпе з метою просвітництва було представлено в межах II Національного конгресу «Актуальні питання перинатальної неврології», що відбулася 17-19 жовтня 2019 р. у Києві.
Клінічний випадок сімейного варіанта метаболічної міопатії
 Дитячий лікар-невролог вищої категорії, співробітниця Українського науково-дослідного інституту медичної реабілітації та курортології МОЗ України (м. Одеса), к. мед. н. Валентина Степанівна Бусова представила цікавий клінічний випадок сімейного варіанта метаболічної міопатії.
Дитячий лікар-невролог вищої категорії, співробітниця Українського науково-дослідного інституту медичної реабілітації та курортології МОЗ України (м. Одеса), к. мед. н. Валентина Степанівна Бусова представила цікавий клінічний випадок сімейного варіанта метаболічної міопатії.
За словами лікаря, пацієнтка А., 9 років, скаржилася на утруднення з виконанням присідань та ходьби по сходах. Дитина від 1-ї вагітності з нормальним перебігом, пологи на 38-му тижні, тривалість пологів – 12 год, безводний період – 4,5 год (води світлі), маса при народженні – 3400 г, зріст – 52 см, оцінка за шкалою Апгар – 8/8 балів. Із народження виникали епізоди неспокою, сиділа при висаджуванні з 6 міс., у 8,5 міс. самостійно не сідала, у 10 міс. стояла біля опори, але відзначалися труднощі зі вставанням з положення навпочіпки, пішла з 12 міс., до 2 років батьки відзначали труднощі з присіданням, після 3 років – з ходьбою по сходах, втім ходьба на далеку відстань була можливою.
При першому огляді невролога було виявлено наступні ознаки:
- збереження інтелекту;
- зниження глоткового рефлексу, помірну слабкість мімічної мускулатури (симптом «вій»), фібриляцію м’язів язика;
- м’язові атрофії переважно у проксимальних відділах;
- слабкість м’язів тулуба: крилоподібні лопатки, гіперлордоз зі збереженою функцією міжреберних м’язів (дівчинка грає на флейті);
- відсутність симптому «ямки»;
- зниження сили в м’язах плечового поясу (4 бали), у проксимальному (3 бали) та дистальному відділі (2 бали) нижніх кінцівок;
- порушення ходи:
- «качина хода»;
- «степаж» (перонеальна, або «півняча», хода – порушення, зумовлене відвислою стопою), більшою мірою зліва;
- утруднення піднімання сходами (угору до 10 сходинок, хода вниз значно ускладнена);
- труднощі з ходьбою навшпиньки, гірше зліва, неможливість ходьби на п’ятах;
- утруднення з підстрибуванням;
- симптом Говерса (ознака м’язової слабкості – щоб піднятися з положення сидячи навпочіпки, пацієнт потребує спершу поставити руки на підлогу та поступово підвестися, спираючись руками на коліна і стегна);
- відсутність значної фізичної втомлюваності (можлива ходьба на далеку відстань).
- зниження глибоких рефлексів із рук, відсутність карпорадіальних, колінних, ахілових рефлексів;
- зниження вібраційної чутливості в ногах до 11 с (І ступінь зниження, що свідчить про залучення товстих чутливих волокон).
Таким чином, анамнез та клінічна картина вказували на ураження периферичного нейрона або безпосередньо м’язів. Диференційні діагнози, які брала до уваги В.С. Бусова, представлено в таблиці.
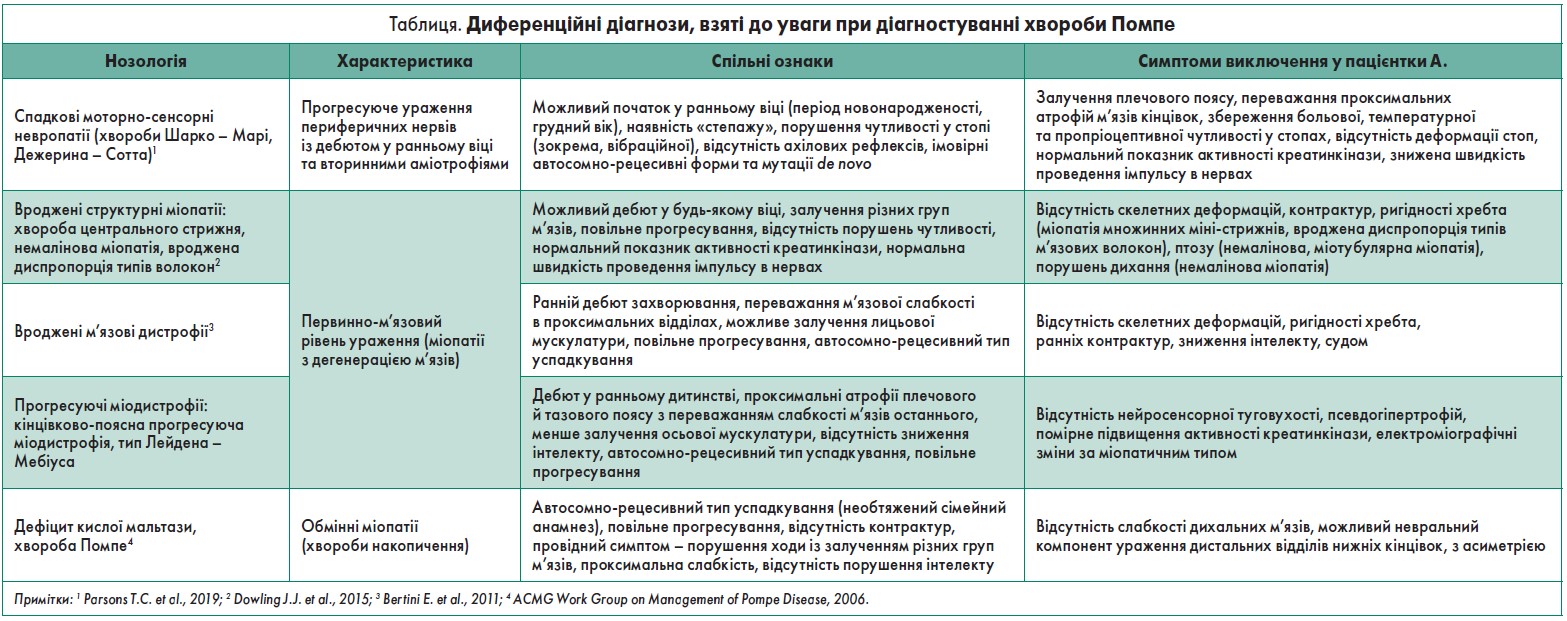
Із метою уточнення діагнозу було проведено:
- Біохімічне дослідження крові: активність креатинфосфокінази (КФК) – 1357 мкмоль/хв×л, активність аспартатамінотрансферази (АСТ) – 298 мкмоль/хв×л, активність аланінамінотрансферази (АЛТ) – 246 мкмоль/хв×л (ознаки міолізу).
- Електроміографія: дані на користь ідіопатичного міопатичного синдрому – зниження амплітуди потенціалів рухових одиниць із поліфазією. Помірно виражені невральні розлади, що не перевищують ступінь м’язового розладу (первинно-м’язовий тип ураження).
У контексті даного клінічного випадку увагу лікаря привернула хвороба Помпе, в основі патогенезу якої лежить мутація гена кислої α-глюкозидази, що призводить до дефіциту даного ферменту, внутрішньоклітинного накопичення глікогену, патологічних змін м’язових волокон, клінічних проявів, втрати здатності до пересування та дихання і, насамкінець, до ранньої смерті (Hirschhom, 2001; Nadine et al., 2012). У 92% дітей із хворобою Помпе у віці до 4 міс. розвиваються кардіомегалія та серцева недостатність (Kishnani et al., 2006). Тому додатково було проведено: ехокардіографію (зниження показника мітрально-септальної сепарації), електрокардіографію (гіпертрофія лівого шлуночка І ступеня, ішемія перетинки) та ультразвукове обстеження органів черевної порожнини (ознаки диспанкреатизму).
Ферментну діагностику хвороби Помпе проводять у лабораторіях ARCHIMED Life Science GmbH Laboratories (Австрія) або Центру орфанних захворювань Національної дитячої спеціалізованої лікарні «Охматдит» МОЗ України (м. Київ). За умови позитивного результату ферментного аналізу зразок відправляють для молекулярно-генетичної діагностики в австрійську лабораторію.
Таким чином, було проведено:
- визначення ферментної активності α‑1,4-глюкозидази в сухих плямах крові (DBS) завдяки підтримці ТОВ «Санофі-Авентіс Україна» – показник становив 0,2 мкмоль/год×л (нормальні значення [N] >2,0);
- генетичний аналіз: дві гетерозиготні мутації с.[1129G>A], [2237G>A] (р.[Gly377Ser], [Trp746Ser]).
У Центрі орфанних захворювань НДСЛ «Охматдит» МОЗ України пацієнтку консультувала завідувачка Центру, к. мед. н. Наталія Олександрівна Пічкур. Після додаткових обстежень було встановлено діагноз: спадкове порушення обміну речовин із групи патологій накопичення глікогену – глікогеноз ІІ типу (хвороба Помпе).
З огляду на автосомно-рецисивний характер успадкування даного захворювання, проведене обстеження членів родини:
1. Пацієнт Н., 10 міс., сиблінг:
- сидить при висажуванні з 8 міс., почав повзати після 9,5 міс., самостійно сідає з 9,5 міс., встає біля опори з 10 міс.;
- помірна м’язова гіпотонія, періостальні рефлекси, сила в ногах збережені;
- активність КФК – 1642,1 мкмоль/хв×л, лактатдегідрогенази (ЛДГ) – 962 мкмоль/хв×л, АЛТ – 234 мкмоль/хв×л, АСТ – 492 мкмоль/хв×л;
- після проведення дослідження DBS рівень активності α‑1,4-глюкозидази – 0,0 мкмоль/год×л (N>2,0);
- генетичний аналіз: дві гетерозиготні мутації с.[1129G>A], [2237G>A] (р.[Gly377Ser], [Trp746Ser]), що підтверджує наявність хвороби Помпе.
2. Батько:
- після проведення дослідження DBS рівень активності α‑1,4-глюкозидази – 1,2 мкмоль/год×л (N>2,0);
- генетичний аналіз: дві гетерозиготні мутації с.[1129G>A], [?] (р.[Gly377Ser], [?]), одна з яких описана при хворобі Помпе та пояснює зниження активності лізосомального ферменту в крові, проте виявлення однієї мутації недостатньо для постановки діагнозу.
3. Мати: після проведення дослідження DBS рівень активності α‑1,4-глюкозидази – 3,6 мкмоль/год×л (N>2,0).
Таким чином, незважаючи на низьку інформованість лікарів, можливість виявлення пацієнтів із хворобою Помпе є реальною та доступною завдяки державній програмі діагностики в Центрі орфанних захворювань НДСЛ «Охматдит» МОЗ України та підтримці ТОВ «Санофі-Авентіс Україна». Раннє виявлення хворих є вирішальним фактором для успішності специфічної замісної терапії ферментами, що забезпечується компанією на благодійних основах.
Можливості оптимізації терапії пацієнтів із хворобою Помпе на основі аналізу клінічного випадку
 Завідувачка відділення неврології молодшого віку Дніпровської дитячої клінічної лікарні № 5, к. мед. н. Ірина Василівна Македонська представила сучасні реальні можливості допомоги пацієнтам із хворобою Помпе. Поширеність цієї патології варіює залежно від етнічної приналежності та географічного розташування. Швидко прогресуюча форма з інфантильним початком зустрічається з частотою 1 : 138 тис. у кавказьких популяціях, 1 : 50 тис. серед китайського населення та 1 : 14 тис. – осіб африканського походження. Поширеність хвороби Помпе є також високою в арабській популяції.
Завідувачка відділення неврології молодшого віку Дніпровської дитячої клінічної лікарні № 5, к. мед. н. Ірина Василівна Македонська представила сучасні реальні можливості допомоги пацієнтам із хворобою Помпе. Поширеність цієї патології варіює залежно від етнічної приналежності та географічного розташування. Швидко прогресуюча форма з інфантильним початком зустрічається з частотою 1 : 138 тис. у кавказьких популяціях, 1 : 50 тис. серед китайського населення та 1 : 14 тис. – осіб африканського походження. Поширеність хвороби Помпе є також високою в арабській популяції.
Незважаючи на незначну поширеність цієї хвороби накопичення, І.В. Македонська навела дані про клінічний випадок із власної практики.
Дитина П., 11 років, діагноз: глікогеноз ІІ типу (хвороба Помпе), атипова форма.
Перинатальний анамнез: дівчинка від 2-ї вагітності, перша вагітність закінчилася пологами, старша дитина здорова. Друга вагітність перебігала на тлі загрози переривання вагітності у 1-й половині, гострої респіраторної інфекції, гестаційної анемії. Пологи на 40-му тижні, спостерігалися слабкість пологової діяльності, передчасне відходження навколоплідних вод зеленого забарвлення. Було проведено ургентний кесарів розтин. Маса дитини при народжені – 3550 г, довжина – 54 см, за шкалою Апгар – 6/8 балів. У зв’язку з асфіксією проводили інтубацію, киснетерапію. До грудей прикладена на 3-тю добу. Була виконана вакцинація проти туберкульозу. Виписана на 8-му добу.
Анамнез захворювання: затримку розвитку мати почала помічати після 6‑7-місячного віку. Приблизно в 1 рік дитина навчилася сидіти, у 13 міс. могла стояти біля опори, у 15 міс. почала ходити з підтримкою, після 2 років ходила самостійно, однак швидко втомлювалася, спотикалася, падала. Почала розмовляти в 3 роки та 2 міс., фразова мова сформувалась у 4,5 років. У 5 років словниковий набір відповідав віку, однак мова була незрозумілою у зв’язку зі слабкістю м’язів артикулярного апарату, макроглосією.
В 11 міс. при огляді невролога було встановлено: міопатичний синдром унаслідок перинатального ураження ЦНС, затримання моторного розвитку. Призначено антихолінестеразні та вітамінні препарати, масаж та лікувальну фізкультуру.
У грудні 2011 р. (3 роки) було діагностовано множинні рабдоміоми мітрального клапана. Оглянута кардіохірургом: рабдоміоми папілярних м’язів мітрального клапана без гемодинамічних порушень, серцева недостатність 0 ступеня.
У липні 2012 р. дитину обстежено у спеціалізованому медико-генетичному центрі Харкова. Додатково до відставання у моторному та мовному розвитку виявлено зміни біохімічного дослідження крові:
- активність АСТ – 340,42 мкмоль/хв×л (N<48);
- активність АЛТ – 340,42 мкмоль/хв×л (N<33);
- активність КФК – 136,18 мкмоль/хв×л (N<228);
- активність ЛДГ – 888,75 мкмоль/хв×л (N<395);
- активність гомоцистеїну – 6,4 мкмоль/хв×л (N<5).
Сформований висновок: наявний дефіцит ферментів фолатного циклу, гіпергомоцистеїнемія, мітохондріальна дисфункція, вроджена м’язова дистрофія; необхідно виключити порушення обміну стеролів, глікогеноз.
Станом на січень 2014 р. (у віці 6 років) у дитини розвинулася гостра респіраторна вірусна інфекція на тлі недифенційованої міопатії. Протягом наступних 5 днів стан прогресивно погіршувався, діагностовано ускладнення – полісегментарна двобічна пневмонія, інтоксикація, дихальна недостатність. У тяжкому стані (атонія, арефлексія, адинамія, відсутність самостійного дихання, відсутність ковтання) госпіталізована в Дніпровську обласну дитячу клінічну лікарню.
Поступово на тлі антибактеріальної терапії та забезпечення оксигенації апаратом штучної вентиляції легень збільшилися показники сили в кінцівках (до 3 балів у верхніх та до 2 балів у нижніх), з’явилися торпідні сухожилкові рефлекси на верхніх кінцівках.
Електроміографія (30.04.2014 р.): не виявлено ознак ураження передніх рогів, функціональна здатність м’язів знижена за первинно-м’язовим типом.
У травні 2014 р. зразок крові для дослідження методом DBS було направлено в лабораторію медичної генетики медико-генетичного відділення НДСЛ «Охматдит» МОЗ України. Через 3 тижні отримано позитивний результат: зниження активності ферменту α-глюкозидази; повторний аналіз підтвердив зниження активності ферменту, що становила 0,15 нмоль/год ×мг білка (N=0,3‑1,2).
На підставі отриманих даних Н.О. Пічкур сформулювала висновок: має місце спадкове порушення обміну речовин із групи глікогенозів (патологій накопичення) – хвороба Помпе (глікогеноз ІІ типу), хронічна форма. Було рекомендоване призначення алглюкозидази альфа у вигляді інфузій в дозі 20 мг/кг один раз на два тижні.
Після підписання батьками інформованої згоди в межах гуманітарної програми компанія Sanofi Genzyme («Санофі») надала алглюкозидазу альфа для ініціації інфузії під керівництвом та контролем завідувача реанімаційного відділення Дніпровської обласної дитячої клінічної лікарні Олександра Ричардовича Лацинського. Терапія переносилася без клінічно значущих побічних явищ. За час специфічного лікування в обласній лікарні стан дівчинки поліпшився, зберігається залежність від апарату штучної вентиляції легень, але збільшився час самостійного дихання вдень, може сидіти у кріслі та грати без апарату до 1,5 год, харчується самостійно, ковтання не порушене, мова поліпшилася; дифузна м’язова гіпотонія, в динаміці показники сили м’язів збільшилися до 3‑4 балів у руках та до 3 балів у ногах; сформувалася контрактура в гомілково-ступневих суглобах; глибокі рефлекси з рук торпідні, з ніг не викликаються. Періодично виникали епізоди агресії, особливо по відношенню до матері, після приймання афобазолу поведінка стабілізувалася.
З метою обмеження ризику повторних інфікувань (хворіла на гострі респіраторні інфекції 1‑2 рази на місяць), поліпшення емоційного стану пацієнтки та батьків після 765 днів перебування в обласній лікарні була розглянута можливість хоспісного нагляду. З 15.12.2015 р. дівчина перебуває в домашніх умовах під наглядом спеціалістів.
У серпні 2016 р. повторно отримані позитивні результати генетичного дослідження з Австрії, діагноз хвороби Помпе підтверджено.
Цікавою є наступна динаміка: у вересні 2017 р. дівчинка могла знаходитися без апарату штучної вентиляції легень до 6 год, вночі дихала виключно за його допомогою, могла сидіти у спеціальному кріслі з підтримкою голови та бавитися іграшками, конструктором до 4 годин два рази на день. Самостійно повзала «по-пластунськи». Зберігалася дифузна гіпотонія. Показники сили в руках – 4 бали, в ногах – 3 бали. Глибокі рефлекси з рук жваві. Однак у листопаді 2017 р. на тлі гострої респіраторної інфекції виник регрес: самостійне дихання не більше за годину на добу, зниження сили.
З огляду на зменшення ефективності замісної терапії ферментами, що може бути зумовлено підвищенням титру антитіл до міозиму, було вирішено провести визначення рівня антитіл класу IgG – результат становив 800. Проведений консиліум за участю Н.О. Пічкур, І.В. Македонської, О.Р. Лацинського; прийняте рішення про поступове підвищення одноразової дози алглюкозидази альфа до 30 мг/кг: 10 інфузій по 25 мг/кг, що становило 500 мг міозиму (10 флаконів), та через 2 тижні – по 30 мг/кг, що становило 600 мг (12 флаконів).
Через місяць стан пацієнтки почав поліпшуватися, повернувся попередній рівень фізичної активності. Нині стан дівчинки стабільний.
Додатково до класичного обстеження на хворобу Помпе І.В. Македонська рекомендує визначати статус перехресно реагуючого імунологічного матеріалу (CRIM), що відображає можливість синтезу деякої кількості ферменту (CRIM-позитивний статус) або повну відсутність ферменту α-глюкозидази (CRIM-негативний статус). Визначення CRIM-статусу дозволяє зробити прогноз та розробити план ведення пацієнта з інфантильною формою хвороби Помпе. На жаль, 20‑25% пацієнтів із ранньою формою є CRIM-негативними; в них екзогенна α-глюкозидаза розпізнається як чужорідна, виробляються нейралізуючі антитіла, що пояснює гіршу відповідь на терапію алглюкозидазою альфа та необхідність призначення адекватної імуномодулювальної терапії до ініціації введення ферменту або через короткий термін після – такі заходи значно поліпшують прогноз. Показано, що CRIM-позитивні хворі є більш незалежними від інвазивної вентиляції легень, демонструють менший ризик високого титру IgG до рекомбінантної α-глюкозидази при замісній терапії ферментами без індукції імунної толерантності (Ball et al., 2012).
Загалом рекомбінантна α-глюкозидаза має деякий потенціал імуногенності, тому титр IgG необхідно визначати один раз на 3 міс. протягом перших 2 років, надалі – щорічно. При виявленні алергічної реакції або інших імуноопосередкованих процесів слід розглянути можливість частішого виявлення титру антитіл. У більшості пацієнтів антитіла до препарату з’являються в перші 3 міс. після початкової інфузії (Schoser et al., 2015).
Стосовно ефективності алглюкозидази альфа І.В. Македонська навела статистичні дані: хороша або помірна відповідь спостерігається у 91% пацієнтів – у вигляді змін показників м’язової сили та у 86% – в плані поліпшення показника функціональної життєвої ємності легень (de Vries et al., 2012).
Беззаперечними є висновки:
- Ефект терапії рекомбінантною α-глюкозидазою є позитивним щодо виживання та затримки прогресування дихальної недостатності у пацієнтів за умови ранньої ініціації.
- Лікування дітей та дорослих суттєво впливає на якість життя (поліпшення рухливості, зменшення потреби в респіраторній підтримці).
- Ранній початок терапії є критичною умовою для оптимізації наслідків хвороби (van der Ploeg et al., 2007).
Підготувала Маргарита Марчук
Тематичний номер «Неврологія, Психіатрія, Психотерапія» № 4 (51) грудень 2019 р.