


16 червня, 2020
Семейные онкологические синдромы и гены наследственного рака
Когда-нибудь, в далеком будущем, слияние генетики и медицины все же произойдет.
Возможно, тогда врач сможет позвать на консультацию своего приятеля-генетика.
Thomas Morgan, лауреат Нобелевской премии 1933 г.
.jpg) Семейный и наследственный рак составляют не менее 20% от всех злокачественных опухолей человека, однако в клинической практике они распознаются реже. Понимание механизмов наследственного канцерогенеза позволяет не только эффективно лечить онкологического больного, но и предупредить у него появление второй (метахронной) опухоли в новом органе. С помощью превентивных диагностических и лечебных мероприятий можно также предотвратить развитие рака у здорового носителя патологической мутации, переданной ему по наследству.
Семейный и наследственный рак составляют не менее 20% от всех злокачественных опухолей человека, однако в клинической практике они распознаются реже. Понимание механизмов наследственного канцерогенеза позволяет не только эффективно лечить онкологического больного, но и предупредить у него появление второй (метахронной) опухоли в новом органе. С помощью превентивных диагностических и лечебных мероприятий можно также предотвратить развитие рака у здорового носителя патологической мутации, переданной ему по наследству.
Следует различать понятия «семейный рак» и «наследственный рак». О семейном раке говорят в тех случаях, когда у нескольких поколений родственников возникают злокачественные опухоли, однако мутации известных генов при этом не обнаруживаются. В таких семьях причиной рака может быть наследование определенного образа жизни (ожирение, курение). Наследственным называют рак, при котором у заболевших представителей нескольких поколений семьи выявляются мутации генов, ассоциированных со злокачественными опухолями.
Технологии обнаружения мутаций генов, входящих в состав разных наследственных синдромов, сегодня доступны. Наиболее удобным генетическим исследованием является метод молекулярного профилирования ДНК, которая присутствует в крови как онкологического больного, так и его ближайших здоровых родственников (Liquid biopsy).
История изучения наследственного рака
Самой известной семьей, в которой «жил рак», была семья Наполеона Бонапарта. Все члены этой семьи – отец Карл Бонапарт, дочь Мария Бонапарт-Боргезе и сын Наполеон Бонапарт – умерли от рака желудка.
 Первое медицинское описание наследственного рака молочной железы в 1866 г. сделал французский врач и антрополог Pierre Broca, у молодой жены которого, а также у четырех поколений женщин этой семьи в молодом возрасте возникали злокачественные опухоли.
Первое медицинское описание наследственного рака молочной железы в 1866 г. сделал французский врач и антрополог Pierre Broca, у молодой жены которого, а также у четырех поколений женщин этой семьи в молодом возрасте возникали злокачественные опухоли.
В 1872 г. Hilario de Gouvea, бразильский офтальмолог, описал случай ретинобластомы, по поводу которой он выполнил энуклеацию глаза маленькому мальчику. Мальчик выжил, вырос, женился и имел детей. У двух его дочерей также развилась ретинобластома, от которой обе умерли.
Медицинский мир проигнорировал эти сообщения, поскольку в то время еще не были сформулированы общие законы наследственности, а учение Gregor Mendel подвергалось жесткой критике.
После того, как были открыты хромосомы и изучен митотический клеточный цикл, в 1911 г. немецкий биолог Theodor Boveri выдвинул хромосомную теорию опухолей и предположил, что существует наследственная предрасположенность к развитию рака. Наблюдая аномалии кариотипа и изменение набора хромосом в злокачественных клетках, Boveri обнаружил, что одни хромосомы стимулируют, а другие тормозят деление клеток. Хромосомная теория рака опередила открытие онкогенов и генов-супрессоров более чем на 60 лет.
 Уже в начале XX века биологам стало ясно, что передается не сам рак, а предрасположенность к развитию опухолей. Karl Bauer в 1928 г. писал, что «не существует наследственной передачи рака в точном смысле этого выражения – речь идет о наследовании склонности различных тканей образовывать опухоли при определенных внешних условиях». Таким образом, Bauer задолго до появления новой науки – эпигенетики – настаивал, что на развитие рака влияют не только генетические, но и внешние факторы.
Уже в начале XX века биологам стало ясно, что передается не сам рак, а предрасположенность к развитию опухолей. Karl Bauer в 1928 г. писал, что «не существует наследственной передачи рака в точном смысле этого выражения – речь идет о наследовании склонности различных тканей образовывать опухоли при определенных внешних условиях». Таким образом, Bauer задолго до появления новой науки – эпигенетики – настаивал, что на развитие рака влияют не только генетические, но и внешние факторы.
Через 60 лет после обнародования хромосомной теории генетик Alfred Knudson, работавший в 1970 г. в онкологическом центре Хьюстона (сегодня Andersen Cancer Center), на основании статистических расчетов данных, полученных из историй болезни пациентов с ретинобластомой, предложил свою теорию развития наследственных опухолей. Новая парадигма получила название теории двойного удара. Knudson доказал, что не существует отдельных генов наследственного рака, а для развития опухоли необходимы две последовательные мутации – в онкогене и гене-супрессоре. При генетической предрасположенности к раку клетка должна содержать одну зародышевую и одну соматическую мутацию, хотя такое сочетание является достаточно редким событием. Наличие зародышевой мутации, передающейся по наследству, объясняет ранний возраст развития наследственных опухолей.
 Герминогенные мутации, возникающие в зародышевых клетках и передающиеся по наследству, имеются не только в раковых, но и во всех нормальных клетках организма. Соматические онкогенные мутации возникают и наследуются только раковыми клетками. Соматические мутации отсутствуют в нормальных клетках и не передаются потомству.
Герминогенные мутации, возникающие в зародышевых клетках и передающиеся по наследству, имеются не только в раковых, но и во всех нормальных клетках организма. Соматические онкогенные мутации возникают и наследуются только раковыми клетками. Соматические мутации отсутствуют в нормальных клетках и не передаются потомству.
Первый ген-супрессор, мутация которого связана с повышенным риском развития наследственного рака молочной железы, был открыт Mary-Claire King в 1988 г. и назван BRCA1 (аббревиатура от breast cancer). Ген был клонирован Mark Skolnick, основателем компании Myriad Genetics. Сегодня известны два гена – BRCA1 и BRCA2, продолжается поиск гена BRCA3. Функциональной ролью генов BRCA1/2 является контроль клеточного цикла и репарации ДНК.
С момента открытия гена BRCA началась новая эра изучения наследственного рака.
Рак молочной железы в составе наследственных онкологических синдромов
Сегодня известно, что мутации BRCA1/2 отвечают за повышенный риск развития рака молочной железы, яичника, маточной трубы, грудной железы у мужчин, поджелудочной и предстательной желез. Пенетрантность этих генов (частота развития рака при мутации гена) достаточно высока – достигает 80%. Например, у Angelina Jolie индивидуальный генетический риск развития рака молочной железы составил 87%.
Выявлены тысячи мутаций генов BRCA1/2, но не все они являются клинически значимыми. Только наиболее важные мутации (185delAG, 5382insC и 6174delT) приводят к повышению риска развития наследственного рака, однако частота этих мутаций невелика и составляет в среднем 1 на 1000 женщин общей популяции.
С появлением технологий молекулярного анализа в результате проведенных онкоэпидемиологических исследований были обнаружены другие пенетрантные гены, мутации которых отвечают за развитие почти 80% всех случаев наследственного рака молочной железы: TP53, PTEN, STK11, CDH1, PALB2, CHEK2, ATM, NBN, BARD1, BRIP1, RAD51C. Оказалось, что эти гены ассоциированы также с наследственным раком других локализаций, который входит в состав многочисленных наследственных синдромов. Характерными примерами таких ассоциаций являются синдромы Р53 (Ли – Фраумени), PALB2, CHEK2 и PTEN (Cowden), при которых наследственный рак молочной железы может сочетаться с раком яичника, предстательной, поджелудочной, щитовидной желез, саркомами, опухолями головного мозга, раком тела матки, толстой кишки и желудка (табл. 1).
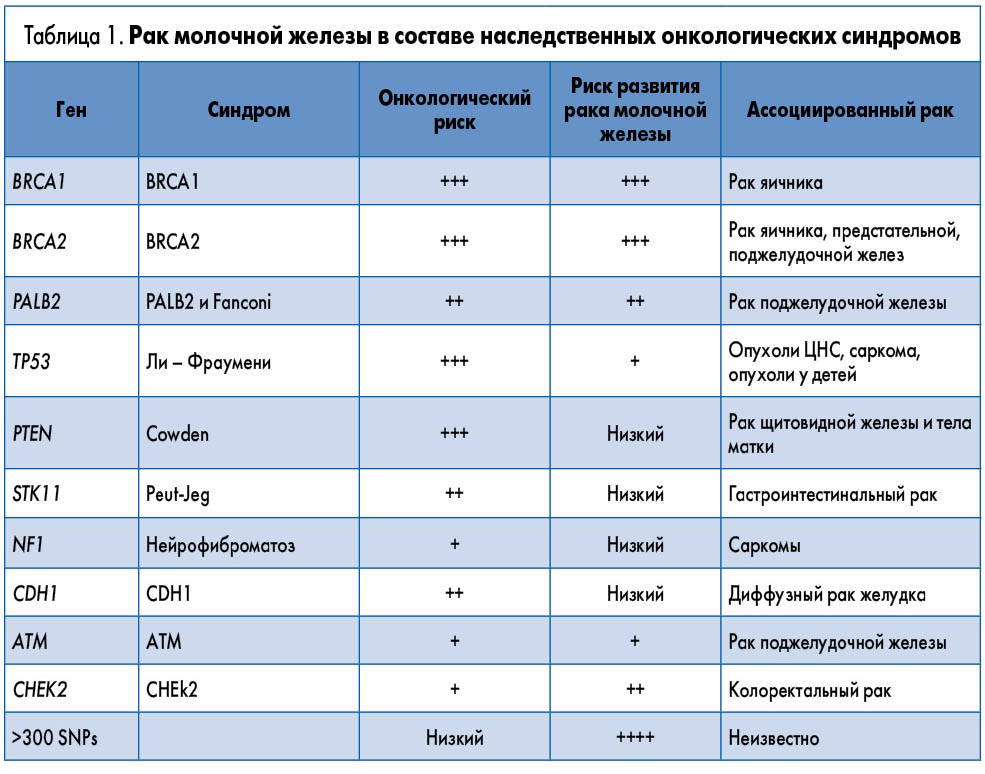
Рак молочной железы в составе синдрома Р53 (Ли – Фраумени)
Фредерик Ли и Джозеф Фраумени описали наследственный синдром, характеризующийся образованием многочисленных синхронных и метахронных опухолей с разнообразными онкологическими ассоциациями. Эти ассоциации включают:
- дольковый рак молочной железы, рак желудка;
- рак молочной железы, саркому мягких тканей, опухоли центральной нервной системы, рак надпочечника, лейкоз, рак предстательной железы;
- рак молочной железы, гамартому, рак щитовидной железы, слизистой оболочки полости рта, эндометрия, опухоли головного мозга;
- рак грудной железы у мужчин, рак поджелудочной железы, желчного пузыря, глотки, желудка, меланому, рак предстательной железы.
Особенностью этих опухолей является раннее развитие заболевания, семейный анамнез и наличие мутации гена р53. Следует подчеркнуть, что канцерогенез этого наследственного синдрома не связан с мутацией BRCA1/2, что имеет значение при дифференциальной диагностике.
Разработан алгоритм профилактического наблюдения здоровых членов семьи, носителей наследственной мутации р53. Учитывая риск развития карциномы надпочечника, регулярно, с детского возраста следует проводить ультразвуковое исследование брюшной полости и определять уровень кортизола мочи. При положительном результате генетического теста с 18 лет необходимо проводить ежегодные обследования молочной железы и обсудить вопросы профилактической мастэктомии. Из-за угрозы развития саркомы мягких тканей и костей необходимо ежегодно проводить магнитно-резонансную томографию (МРТ) всего тела. В связи с повышенным риском развития опухоли ЦНС ежегодно выполняется МРТ головного мозга. С 25 лет каждые два года необходимо проводить колоноскопию и ежегодно – дерматоскопию всего тела. Каждые 3-4 месяца следует выполнять анализ крови (включая уровень лактатдегидрогеназы).
Онкологическим больным с синдромом Ли – Фраумени следует избегать лучевой терапии и применения генотоксических противоопухолевых препаратов.
Рак молочной железы в составе синдромов PALB2, CHEK2 и PTEN
Наследственные мутации гена PALB2 связаны с 30-60% риском развития наследственного рака молочной железы. Наиболее высока ассоциация этой карциномы с раком яичника и поджелудочной железы. Имеется также высокий риск развития рака грудной железы у мужчин.
Нет доказательств повышенного риска развития рака толстой кишки или простаты и общего повышения риска рака других локализаций при мутации PALB2. Изучается ассоциация мутации с риском возникновения рака желудка.
Наследственные мутации гена CHEK2 ассоциированы с повышением на 30% риска развития аденокарциномы молочной железы и колоректального рака.
Потеря гена-супрессора PTEN (синдром Cowden) повышает риск развития рака молочной железы в течение жизни на 25‑50%. Для этого синдрома характерны поражение слизистых оболочек (99%), гамартомные полипы кишечника (60-90%), немедуллярный рак щитовидной железы, рак эндометрия, ганглиоцитома мозжечка у взрослых (болезнь Лермита – Дюкло), карцинома почек.
Наследственный рак молочной железы и рак желудка
Особый интерес представляет онкологическая ассоциация наследственного рака молочной железы и рака желудка, которая возникает при мутации гена CDH1, кодирующего белок E-cadherin. Этот ген участвует в поддержании эпителиальной архитектуры, нарушает клеточную адгезию, клеточную организацию и полярность.
Мутация CDH1 ассоциирована с диффузным раком желудка (linitis plastica), пожизненный риск развития которого при этой аномалии составляет 50-80%, а также с дольковым раком молочной железы, риск возникновения которого у женщин равняется 40%. При мутации CDH1 комбинированный риск развития синхронного и метахронного рака молочной железы и рака желудка у женщин составляет почти 80%. У носителей мутации повышен также риск формирования колоректального рака.
Особенностью синдрома CDH1 является очень высокая вероятность развития перстневидноклеточного диффузного рака желудка, обнаружить который с помощью профилактических серийных эндоскопий и гастробиопсий практически невозможно из-за локальных очагов поражения. Патология проявляется клинически на стадии инфильтративной диффузной карциномы. В связи с этим здоровым носителям патологической мутации гена CDH1 рекомендуется выполнение профилактической гастрэктомии.
После выполнения профилактической гастрэктомии у женщин с 30-летнего возраста проводится скрининг долькового рака молочной железы и у пациентов обоих полов от 40 лет – скрининг рака толстой кишки.
Очевидно, что не все здоровые люди подлежат тестированию на наличие мутации CDH1. Генетическое исследование у членов семей, включая 1-ю и 2-ю степени родства, проводится по определенным показаниям. К ним относятся два случая рака желудка в семье, независимо от возраста (один подтвержденный диффузный рак желудка), один случай диффузного рака желудка в возрасте младше 40 лет, наличие метахронного диффузного рака желудка и долькового рака молочной железы у одного члена семьи в возрасте младше 50 лет, двусторонний дольковый рак молочной железы у одного члена семьи, два или более случаев долькового рака молочной железы в возрасте младше 50 лет, а также обнаружение перстневидных клеток при выполнении гастробиопсии.
Генетическое исследование
В 1980-х годах социологи, феминистки и некоторые онкологи выражали обеспокоенность по поводу быстрой коммерциализации генетических тестов. Причинами опасений были невозможность предотвратить наследственную болезнь, тот факт, что наследственный рак не поддается регулированию со стороны государственной политики, а также необходимость психологической оценки пациентов перед проведением генетического исследования. Последующий опыт большинства стран показал, что психологические последствия генетического тестирования не приводят к увеличению беспокойства или депрессии.
Генетическое тестирование широко распространено среди лиц европеоидной расы в Северной Америке, Евросоюзе, Австралии и Израиле. Из-за конкуренции за ограниченные ресурсы здравоохранения генетическое тестирование не распространено в Азии, Африке и Южной Америке.
Особенностями и преимуществами генетического теста является то, что генетические аномалии присутствуют задолго до появления клинических симптомов рака и остаются без изменений на протяжении всей жизни. Эти мутации имеются у всех членов семьи и их можно отследить у всех родственников. Генетическое тестирование имеет большое значение для предгестальной и пренатальной оценки онкологических рисков.
Кандидаты для генетического исследования
Очевидно, что не следует проводить генетическое тестирование у каждого онкологического пациента. Поиск генов наследственного рака следует начинать только после генетического консультирования и изучения семейного онкологического анамнеза. Заподозрить семейный (наследственный) характер рака можно в следующих клинических ситуациях:
- рак в раннем возрасте (например, рак толстой кишки в 20-летнем возрасте);
- более одного типа рака у одного и того же человека (например, рак молочной железы и яичника у одной женщины);
- рак, возникающий в обоих парных органах (глаза, почки, молочные железы);
- одинаковые опухоли у детей из одной семьи (саркома у брата и сестры);
- рак, возникающий в нескольких поколениях (например, рак у деда, отца и сына);
- множественные случаи одного и того же типа рака (особенно если это необычный или редкий тип опухоли, например, ретинобластома);
- рак грудной железы у мужчины.
Технологии генетического тестирования
Обнаружение известных одиночных мутаций ДНК (старая, простая и хорошо налаженная технология) уже не может удовлетворять потребности клиницистов. Поскольку большинство случаев наследственного рака входит в состав многочисленных онкологических синдромов, изолированное тестирование одного гена не только имеет низкую прогностическую ценность, но и может привести к серьезным негативным последствиям из-за ошибочного диагноза.
Например, у молодой женщины с анамнезом семейного рака молочной железы отрицательный результат теста на мутацию BRCA1/2 еще не может исключить наследственный характер заболевания. Эта пациентка может оказаться носителем одного из многих мутантных генов (TP53, PTEN, STK11, CDH1, PALB2, CHEK2, ATM, NBN, BARD1, BRIP1, RAD51C, CDH1), ассоциированных не только с раком молочной железы, но и других локализаций (желудок, кишечник, тело матки, яичник, ЦНС). Поэтому при отрицательном результате генетического исследования у пациента сохранится высокий риск развития второй наследственной опухоли de novo, а у членов семьи сформируется ложное чувство безопасности. Неполноценное и неадекватное обследование компрометирует идею генетического скрининга.
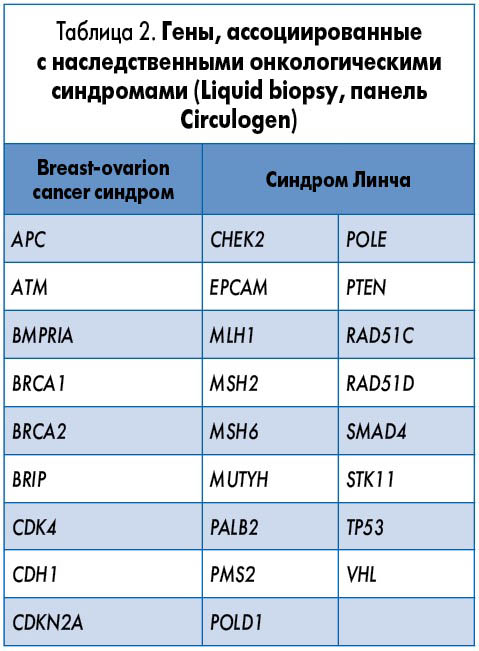 Сегодня клиницистам стали доступны новые молекулярные технологии генетического тестирования, позволяющие идентифицировать в одной панели все известные мутации генов наследственного рака (табл. 2). Наиболее перспективным методом является прямое секвенирование с определением всех геномных вариаций в циркулирующей в крови ДНК (Liquid biopsy). Эта технология доступна в Украине.
Сегодня клиницистам стали доступны новые молекулярные технологии генетического тестирования, позволяющие идентифицировать в одной панели все известные мутации генов наследственного рака (табл. 2). Наиболее перспективным методом является прямое секвенирование с определением всех геномных вариаций в циркулирующей в крови ДНК (Liquid biopsy). Эта технология доступна в Украине.
Отрицательный или неопределенный результат теста
Даже при наличии семейного анамнеза и явных клинических признаков наследственного рака результат генетического исследования может оказаться отрицательным. Есть несколько причин этого.
Возможно, в обследуемой семье действительно нет мутаций и наследственной предрасположенности к раку (семейный, а не наследственный характер рака). Возможно, был протестирован не тот человек. Важно помнить, что первым в семье генетическому исследованию подвергается онкологический больной и только после положительного результата теста обследуют родственников 1-й и 2-й степени родства. При отрицательном результате генетического теста у больного раком (при условии проведения полного молекулярного профилирования) тестирование членов семьи не проводится.
Причиной ложноотрицательного результата исследования чаще всего является несовершенная технология молекулярной визуализации (поиск мутаций одного гена с помощью PCR или FISH), а также, возможно, использование ограниченной панели, в которой нет мутированного гена.
Даже при проведении полного генетического профилирования с оценкой результатов с помощью методов биоинформатики результат теста может оказаться отрицательным, поскольку причинная наследственная мутация находится в небелковой кодирующей области генома.
Наконец, при проведении молекулярных тестов возможны обычные человеческие ошибки.
У части обследуемых могут быть обнаружены мутации, которые не ассоциированы с риском развития наследственного рака. У большинства, как ожидается, эти мутации не будут нести онкогенный потенциал и окажутся безвредными. Такое состояние называется вариантом неопределенного клинического значения (Variant of Uncertain clinical Significance – VUS). От до 2 до 10% людей общей популяции будут иметь VUS.
Выводы
Несмотря на отсутствие международных стандартов, общепризнанной классификации мутаций, сложности с отчетностью и интерпретацией данных, генетическое исследование наследственного рака является быстро развивающимся направлением клинической онкологии.
Тесная интеграция между генетиками и онкологами уже сегодня позволяет успешно влиять на принятие многих клинических решений. Генетическое тестирование у онкологического больного позволяет определить риск развития метахронного рака в другом органе (яичник, матка, кишечник), что открывает возможности для проведения эффективной вторичной профилактики. Генетическое исследование может уточнить риск возможного рецидива опухоли и в некоторых случаях содействовать реализации идеи персонифицированной терапии, основанной на молекулярном профилировании мутаций генов наследственного рака (например, использование PARP-ингибиторов при мутации BRCA1/2).
Для родственников больного, унаследовавших патологическую мутацию, преимущества генетического тестирования заключаются в первичной профилактике рака с помощью индивидуального алгоритма наблюдения и скрининга, а также выполнении в некоторых случаях профилактических операций на органах-мишенях (превентивная мастэктомия, гастрэктомия, колонэктомия).
Следует помнить, что частота наследственного рака в общей популяции больных достаточно высока, а генетическое обследование онкологических пациентов и членов семей с отягощенным анамнезом позволяет улучшить результаты лечения этих пациентов.
За рамками статьи остались множество наследственных онкологических синдромов, проблему которых можно обсудить в следующих номерах.
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 2 (63) 2020 р.