


23 липня, 2020
Спадковий рак молочної залози та яєчника: значення діагностики для профілактики та лікування
.jpg) Спадковий рак молочної залози (РМЗ) та рак яєчника (РЯ) є найпоширенішим спадковим пухлинним синдромом, тобто генетичним захворюванням, яке значно підвищує ризик розвитку злоякісних пухлин. Генетичний скринінг осіб групи ризику необхідний насамперед з метою виявлення здорових носіїв спадкових мутацій для забезпечення ефективної профілактики та ранньої діагностики онкозахворювання. Виявлення спадкових мутацій в онкологічних пацієнтів також дуже важливе, оскільки впливає на тактику їх подальшого лікування.
Спадковий рак молочної залози (РМЗ) та рак яєчника (РЯ) є найпоширенішим спадковим пухлинним синдромом, тобто генетичним захворюванням, яке значно підвищує ризик розвитку злоякісних пухлин. Генетичний скринінг осіб групи ризику необхідний насамперед з метою виявлення здорових носіїв спадкових мутацій для забезпечення ефективної профілактики та ранньої діагностики онкозахворювання. Виявлення спадкових мутацій в онкологічних пацієнтів також дуже важливе, оскільки впливає на тактику їх подальшого лікування.
Для діагностики принциповим є використання методу секвенування нового покоління (NGS), що дозволяє ідентифікувати усі можливі мутації в генах, асоційованих із синдромом спадкового РМЗ та РЯ. У цьому дослідженні 30-генна NGS‑панель дає змогу виявити спадкові мутації у 33% (20/61) пацієнтів з РМЗ або РЯ, тоді як метод полімеразної ланцюгової реакції (ПЛР) – лише у 6% (6/108) пацієнтів в аналогічній вибірці. Таким чином, методом ПЛР можна виявити лише близько 15% спадкових мутацій, отже, він неефективний для скринінгу.
Біологія спадкового раку
Сімейні форми раку спостерігають з давніх часів, однак тільки нещодавно стали відкривати причини підвищеної частоти випадків онкологічних захворювань у деяких сім’ях. У 1994 р. було виявлено перший ген, відповідальний за розвиток РМЗ – BRCA1 (від Breast Cancer 1; Y. Miki et al., 1994), а через рік – ще один, BRCA2 (R. Wooster et al., 1995). Ці гени є класичними онкосупресорами та відіграють важливу роль у процесах репарації, тобто виправленні ушкоджень ДНК, і запускають один із механізмів канцерогенезу (K. Gudmundsdottir, A. Ashworth, 2006; S.A. Narod, W.D. Foulkes, 2004).
Існують два принципово різних механізми канцерогенезу. Перший полягає у виникненні драйверних мутацій в онкогенах, що є стимуляторами росту та розмноження клітин. Активуюча мутація в одному алелі онкогена призводить до зміни функціонування клітини і посилення росту та проліферації, навіть за відсутності мутації в іншому алелі. Мутації в онкогенах спричиняють розвиток спорадичного раку і в більшості випадків є соматичними, тобто такими, що виникають в клітинах тіла (не в гаметах), проявляються тільки у даному організмі та не успадковуються. Біологічною причиною того, що ці мутації не можуть бути спадковими, є те, що навіть їх гетерозиготне носійство (тобто присутність одного мутантного алеля і одного нормального алеля «дикого» типу – WT) призводить до порушення функціонування клітини та внутрішньоутробного розвитку, тому організми зі спадковими мутаціями в онкогенах мають гинути на ранніх етапах ембріогенезу. Тим не менше було показано, що в 0,5% пацієнтів із раком легені, які не курили, виявляють спадкову мутацію T790M в онкогені EGFR (N. Girard et al., 2010). Біологічним поясненням такого явища може бути те, що ця мутація є активуючою тільки в комбінації з іншими мутаціями EGFR, а не сама по собі.
Однак більшість спадково зумовлених онкологічних захворювань пов’язані з іншим шляхом канцерогенезу, ключову роль у якому відіграють гени – супресори пухлинного росту. На сьогодні відома велика кількість генів-онкосупресорів, мутації в яких підвищують ризик розвитку певних видів раку. Найбільш клінічно значущими з них є BRCA1, BRCA2, TP53, PALB2, PTEN, CHEK2, MLH1, MSH2 та ін. Загальний принцип цього шляху канцерогенезу полягає в тому, що спадкова мутація інактивує один алель гена, котрий тим чи іншим шляхом запобігає злоякісній трансформації клітини. Друга, нормальна копія гена компенсує цю втрату, зберігаючи нормальне функціонування клітин та організму в цілому. Однак у клітинах постійно виникають мутації, і є висока вірогідність інактивації другої копії гена, що може призвести до ініціації росту пухлини. У випадку генів BRCA1 та BRCA2, які кодують білки системи репарації дволанцюгових ДНК, друга мутація призводить до накопичення помилок у ній. Здебільшого активуються гени контролю клітинного циклу, які блокують подальший ріст клітин із генетичними аномаліями й індукують програмовану загибель клітини – апоптоз. Але мутації можуть виникнути і в генах регуляції клітинного циклу та ініціації апоптозу (наприклад, у ТР53), таким чином перешкоджаючи загибелі клітини та сприяючи подальшому підвищенню генетичної нестабільності. На цьому етапі залишається ще друга «лінія оборони» – система протипухлинного імунітету, котра може виявити та знищити аномальні клітини. Однак враховуючи частоту виникнення нових мутацій, кількість клітин в організмі та тривалість життя людини, вірогідність розвитку пухлини у носіїв мутацій у генах-онкосупресорах, незважаючи на всі захисні механізми, є дуже високою.
Епідеміологія спадкового РМЗ та РЯ
Частота носійства мутацій, асоційованих із високим ризиком розвитку РМЗ та РЯ, є доволі високою і підвищується за наявності особистого та сімейного онкологічного анамнезу. Найпоширенішими є мутації в генах BRCA1 та BRCA2, але носії цих мутацій становлять не більше половини популяції підвищеного онкологічного ризику. У решти цієї когорти ризик розвитку раку підвищений за рахунок мутацій в інших генах, поширеність яких варіює залежно від популяції (D. Ford et al., 1998; S. Thomas et al., 2002; S.A. Narod, W.D. Foulkes, 2004).
Який же реальний вплив зазначених мутацій на ризик розвитку пухлини? Щодо генів BRCA1 і BRCA2, то мутації в них підвищують ризик розвитку РМЗ до 87%, а РЯ – до 50% протягом життя (S. Chen, G. Parmigiani, 2007; M.C. King et al., 2003; рис. 1). Також у пацієнток з BRCA-асоційованим РМЗ значно підвищується ризик розвитку другої пухлини (K.E. Malone et al., 2010; К.A. Metcalfe et al., 2005; рис. 2).
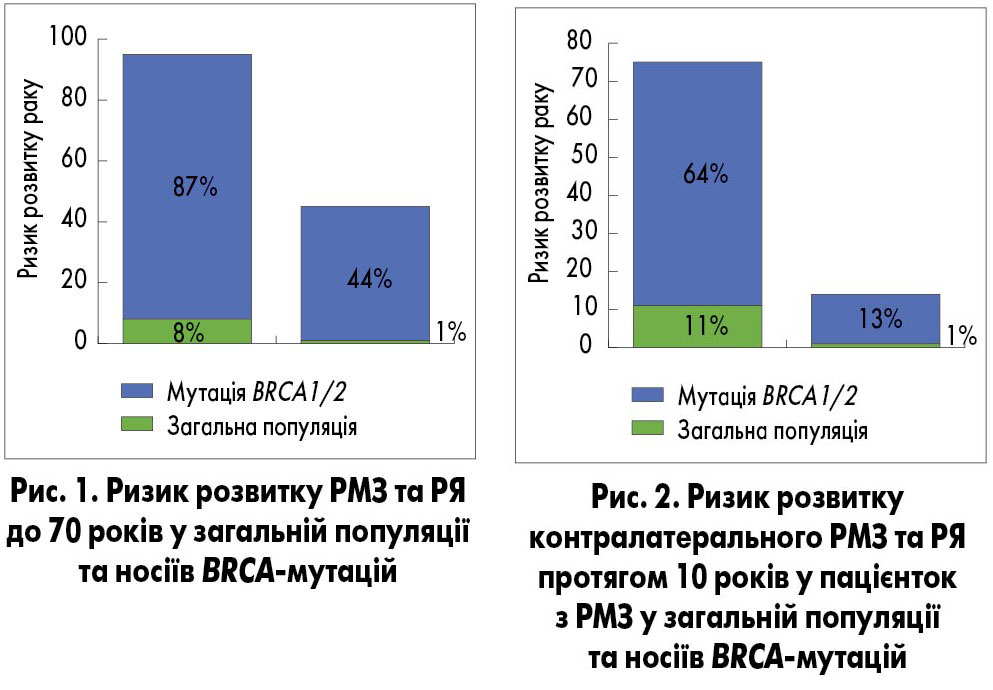
Генетичне обстеження населення головним чином спрямоване на виявлення групи ризику, для якої необхідним є інтенсивний скринінг з метою запобігання або раннього виявлення онкологічного захворювання.
Тактика виявлення носіїв мутацій
Показання до тестування на спадкову схильність до РМЗ та РЯ представлені багатьма міжнародними організаціями, в тому числі Національною онкологічною мережею США (NCCN).
Критерії NCCN для призначення тестування на виявлення генів, асоційованих із високим ризиком розвитку РМЗ і РЯ
1. Наявність мутації в одному з генів у кровного родича.
2. Негативний результат попереднього обмеженого дослідження (наприклад, ПЛР BRCA1/2) за наявності критеріїв для тестування.
3. Персональний онкологічний анамнез:
3.1. РМЗ при одному з критеріїв:
3.1.1. діагностовано у віці ≤45 років;
3.1.2. діагностовано у віці 46-50 років та:
- невідомий сімейний онкологічний анамнез або
- другий РМЗ в будь-якому віці або
- ≥1 близький родич із РМЗ, РЯ, раком підшлункової чи передміхурової залози (≥7 за шкалою Глісона) у будь-якому віці або
3.1.3. потрійний негативний РМЗ, діагностований у віці ≤60 років;
3.1.4. діагностований у будь-якому віці та:
- належність до євреїв-ашкеназі або
- ≥1 близький родич із РМЗ у віці ≤50 років або РЯ, раком підшлункової чи метастатичним раком передміхурової залози в будь-якому віці або
- ≥2 випадків РМЗ у близьких родичів;
3.1.5. рак грудної залози у чоловіків у будь-якому віці.
3.2. Епітеліальний РЯ (переважно High-grade серозний), включно з раком маткових труб і первинним перитонеальним раком, в будь-якому віці.
3.3. Екзокринний рак підшлункової залози в будь-якому віці.
3.4. Метастатичний рак передміхурової залози в будь-якому віці.
3.5. Рак передміхурової залози (≥7 за шкалою Глісона) та:
3.5.1. належність до євреїв-ашкеназі або
3.5.2. ≥1 близький родич із РМЗ у віці ≤50 років або РЯ, раком підшлункової чи метастатичним раком передміхурової залози в будь-якому віці або
3.5.3. ≥2 випадків РМЗ або рак передміхурової залози у близьких родичів у будь-якому віці.
3.6. У пухлині виявлена мутація, яка має клінічне значення, якщо є герміногенною (спадковою).
3.7. Для визначення терапевтичних можливостей, наприклад при HER2-негативному РМЗ.
4. Сімейний онкологічний анамнез:
4.1. особа з онкологічним захворюванням або без нього з родичем 1-2-го ступеня спорідненості, що відповідає переліченим вище критеріям (окрім останнього);
4.2. особа з онкологічним захворюванням або без нього, що не відповідає переліченим критеріям, але має вірогідність мутації BRCA1/2 ≥5%, визначену за моделями ризику (BRCAPro, Pennll та ін.)
Отже, дослідження рекомендується особам з обтяженим сімейним анамнезом, пацієнтам молодого віку, з первинно-множинними пухлинами та всім пацієнтам з РЯ та раком підшлункової залози.
Методи виявлення спадкових мутацій, асоційованих із високим ризиком розвитку РМЗ і РЯ, та порівняння їх ефективності
Спектр генів та різновидів мутацій, що підвищують ризик розвитку РМЗ і РЯ, є надзвичайно широким. Тільки в генах BRCA1 та BRCA2 на сьогодні описано >1800 (https://arup.utah.edu/database/BRCA/Home/BRCA1_landing.php) та >2100 (https://arup.utah.edu/database/BRCA/Home/BRCA2_landing.php) варіантів, близько 94,5% з них є патогенними. Але є мутації, які зустрічаються в популяціях частіше за інші. Так, найчастішими є дві мутації в гені BRCA1 (185delAG і 5182insC) та одна в гені BRCA2 (6174delT). Через досить високу частоту порівняно з іншими ці мутації були описані найпершими і спочатку становили близько 90% усіх описаних спадкових мутацій, асоційованих із РМЗ і РЯ. Це призвело до того, що в деяких країнах, зокрема в Україні, широкого застосування набули ПЛР-тести, які виявляють ці та деякі інші мутації. Але в процесі накопичення даних виявляли все нові варіанти, тому частка зазначених варіантів у всіх носіїв спадкових мутацій поступово зменшувалася і на кінець 2000-х років становила лише 20-30% (L. Zhang et al., 2009).
У Медичній лабораторії CSD було проведено ретроспективне порівняння ефективності застосування ПЛР та NGS для виявлення спадкових мутацій у пацієнтів з РМЗ і РЯ. Для цього було відібрано когорту пацієнтів з РМЗ або РЯ, яким проводилось NGS-дослідження з використанням 30-генної панелі, та випадкову когорту пацієнтів з РМЗ або РЯ, яким було виконано ПЛР-тестування.
У ПЛР-когорту увійшло 108 пацієнток, 103 з яких мали діагноз РМЗ, а 5 – РЯ. Середній вік пацієнток становив 47,5 року. В NGS-когорту увійшла 61 пацієнтка, з яких 49 мали діагноз РМЗ, а 12 – РЯ. Середній вік у NGS-когорті становив 45,5 року.
При ПЛР-дослідженні визначали 5 мутацій у гені BRCA1 (4153delA, 5382insC, 185delAG, 3875del4, 2080delA(ins A)) та 1 мутацію в гені BRCA2 (6174delT). NGS-панель включала повноекзонне секвенування 30 генів (APC, ATM, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A (p14ARF), CDKN2A (p16INK4a), CHEK2, EPCAM, GREM1, MITF, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, POLE, PTEN, RAD51C, RAD51D, SMAD4, STK11, TP53).
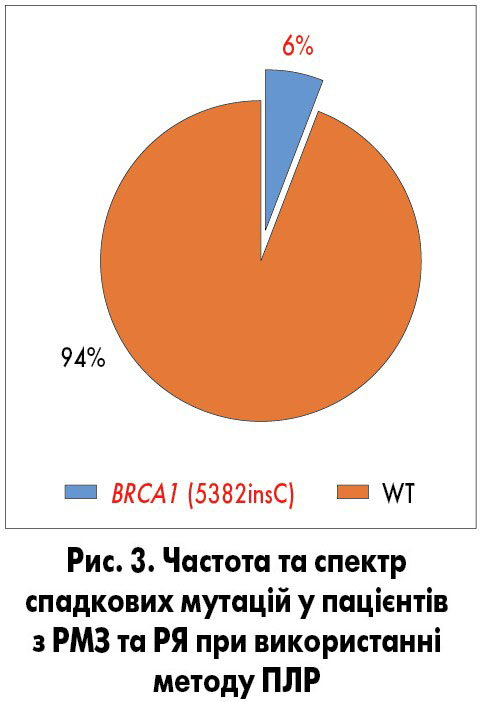 У ПЛР-когорті мутацію було виявлено у 6 пацієнток з РМЗ. У всіх цих пацієнток була визначена мутація BRCA1 5182insC. Загальна частота мутацій склала 6% (рис. 3).
У ПЛР-когорті мутацію було виявлено у 6 пацієнток з РМЗ. У всіх цих пацієнток була визначена мутація BRCA1 5182insC. Загальна частота мутацій склала 6% (рис. 3).
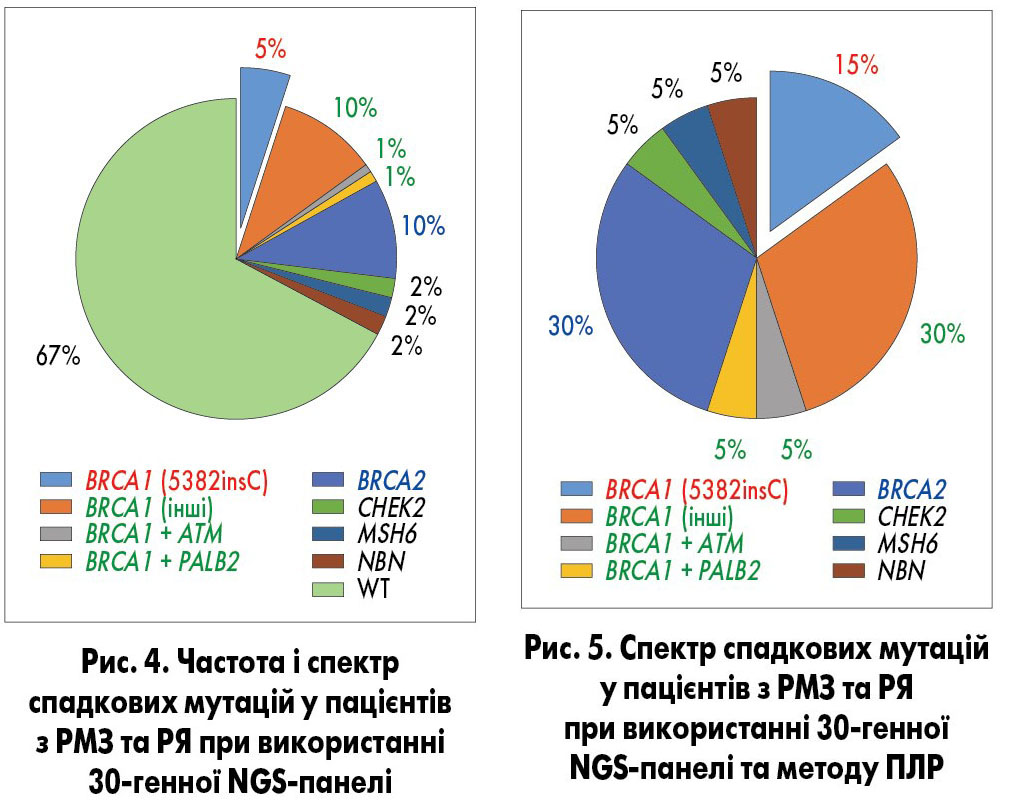
У NGS-когорті мутації було виявлено у 20 пацієнток, у 5 з них було діагностовано РЯ, а у 15 – РМЗ. Серед мутацій, що визначаються методом ПЛР, у NGS-когорті було виявлено лише мутацію BRCA1 5182insC у 3 пацієнток з РМЗ, і її частота склала 5%. Це відповідає частоті мутації в ПЛР-когорті та вказує на те, що когорти є зіставні, а їх порівняння – коректне. Окрім вказаної мутації BRCA1 5182insC, у NGS-когорті було виявлено інші рідкісні мутації BRCA1 – в 10% (6 пацієнтів), BRCA2 – в 10% (6 пацієнтів), CHEK2, MSH6 та NBN1 – по 2% (по 1 пацієнту). У 2 пацієнтів було визначено по дві мутації: BRCA1 + ATM та BRCA1 + PALB2. Загальна частота мутацій у NGS‑когорті склала 33% (рис. 4).
Таким чином, 30-генна NGS-панель виявляє в 5,5 разу більше носіїв мутацій порівняно з ПЛР. При дослідженні NGS-когорти методом ПЛР було б виявлено лише 15% від усіх носіїв спадкових мутацій (рис. 5). Ці результати свідчать про вкрай низьку ефективність і необґрунтованість застосування методу ПЛР для скринінгу мутацій, асоційованих із високим ризиком розвитку РМЗ і РЯ.
Тактика спостереження здорових носіїв спадкових мутацій
Скринінг спадкових мутацій насамперед є важливим для здорового населення з метою запобігання або ранньої діагностики онкологічних захворювань. Після отримання позитивного результату дослідження вибір подальшої тактики визначається індивідуально, спільно лікарем і пацієнтом. Відповідальність за інформування кровних родичів про свій статус мутації та їх потенційний ризик несе сам носій мутації.
Найбільш детально тактика зниження ризику розвитку РМЗ і РЯ розроблена для носіїв мутацій BRCA1/2. Це пояснюється тим, що для вказаних осіб накопичено найбільший масив доказів відносно ефективності тих чи інших процедур.
NCCN наводить основні рекомендації щодо тактики ведення носіїв мутацій, які підвищують ризик розвитку РМЗ і РЯ (табл.). Зокрема процедури зі зниження онкологічного ризику можна розділити на 3 групи: спостереження (скринінг), профілактична хірургія та медикаментозна терапія (https://www.nccn.org). Найбільш вивченими є перші дві тактики, тому більш детально їх розглянемо.
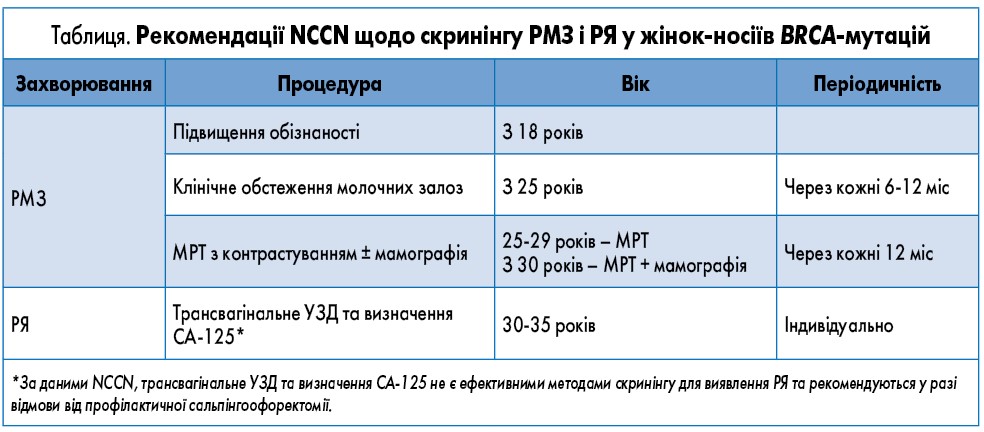
Скринінгові процедури, що підвищують раннє виявлення РМЗ, включають щорічне клінічне обстеження та магнітно-резонансну томографію (МРТ) молочних залоз із 25 років, а після 30 – МРТ у комплексі з мамографією.
Для профілактики РЯ NCCN рекомендує щорічне трансвагінальне ультразвукове дослідження (УЗД) у комплексі з визначенням в крові онкомаркера СА‑125. Однак автори відмічають, що ця тактика не забезпечує істотного зниження ризику РЯ та рекомендується тільки для пацієнток, які відмовилися від профілактичної сальпінгоофоректомії.
Високу ефективність щодо зниження ризику виникнення РМЗ і РЯ продемонстрували профілактичні операції. Так, двобічна профілактична мастектомія знижує ризик розвитку РМЗ на 90%, а сальпінгоофоректомія знижує і ризик розвитку РМЗ на 68% і РЯ на 96% (S.M. Domchek et al., 2010; A. Finch et al., 2006; рис. 6).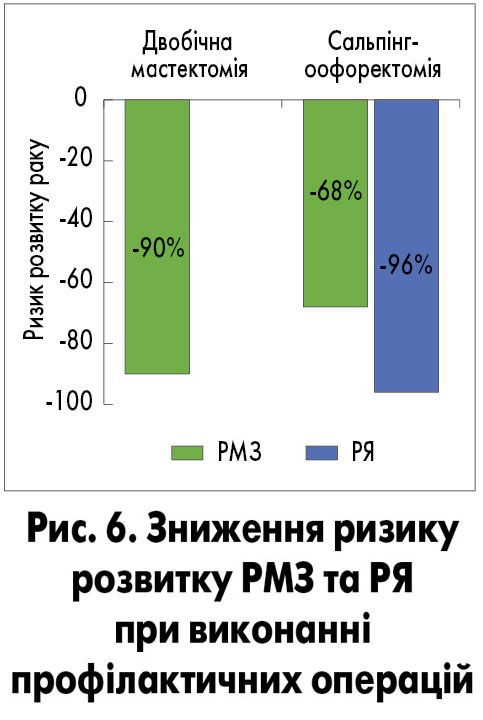
Системне виявлення здорових носіїв спадкових мутацій і чітке дотримання клінічних рекомендацій щодо скринінгових досліджень дуже важливі для зниження захворюваності та підвищення ранньої діагностики РМЗ і РЯ на загальнодержавному рівні.
Значення спадкових мутацій для пацієнтів з РМЗ і РЯ
Окрім того, що інформація про наявність спадкових мутацій важлива для здорових носіїв з метою профілактики онкозахворювань, вона також має істотне значення для пацієнтів з РМЗ і РЯ, оскільки впливає на подальшу тактику терапії.
По-перше, статус мутацій BRCA1/2 важливий для пацієнтів з РМЗ, яким можна виконати органозбережні операції. Було показано, що ризик локальних рецидивів у носіїв мутацій BRCA1/2 при виконанні органозбережної операції в кілька разів вищий, ніж у пацієнтів, яким було виконано мастектомію (Pierce et al., 2010). Крім того, незалежно від виду першої операції, приблизно у половини пацієнтів з BRCA-асоційованим РМЗ протягом 20 років розвивається контралатеральний РМЗ. Таким чином, пацієнтам з локальним і місцевопоширеним BRCA-асоційованим РМЗ рекомендується проведення радикальної операції замість органозбережної, бажано розглянути можливість виконання одномоментної двобічної мастектомії з метою уникнення розвитку контралатерального РМЗ.
По-друге, наявність BRCA1/2 впливає на тактику системної терапії при метастатичному РМЗ. Як було описано вище, гени BRCA1/2 входять в систему репарації ДНК, і в спадково зумовлених пухлинах зазвичай інактивовані обидва алелі гена. Відповідно для таких пацієнтів має бути ефективним застосування агентів, які ушкоджують ДНК (наприклад, препаратів платини), тому що вони будуть призводити до накопичення летальних помилок ДНК у пухлинних клітинах. Результати клінічних досліджень підтвердили це припущення, і часткова або повна відповідь на платиновмісну терапію спостерігається більш ніж у 60% пацієнтів з мутаціями BRCA1/2 (J.M. Lee et al., 2014; K. Alsop et al., 2012; K. Pennington et al., 2014).
Виявлення BRCA-асоційованих пухлин і визначення функції BRCA1/2 у клітині спонукало до пошуку нових терапевтичних мішеней серед білків, що задіяні в системах репарації ДНК. Такі препарати мають блокувати альтернативний шлях відновлення ушкоджень ДНК і таким чином призводити до швидкого їх накопичення та загибелі клітин з неактивним BRCA1/2. Першою групою таких препаратів стали інгібітори полі-АДФ-рибоза-полімерази (PARP), ферменту, який задіяний в репарації одноланцюгових розривів ДНК. Ці препарати продемонстрували свою високу ефективність при BRCA-асоційованих пухлинах і на сьогодні схвалені для терапії пацієнтів з РМЗ і РЯ з мутаціями BRCA1/2.
Таким чином, виявлення здорових носіїв і пацієнтів зі спадково зумовленими пухлинами є надзвичайно важливим для зниження захворюваності та підвищення ефективності лікування РМЗ і РЯ.
Література
- ACOG Practice Bulletin No. 89. Elective and risk-reducing salpingo-oophorectomy. Obstet Gynecol. 2008 Jan; 111(1): 231-41.
- Alsop K., Fereday S., Meldrum C. et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. Journal of Clinical Oncology. 2012; 30(21): 2654‑63.
- Brose M.S., Rebbeck T.R., Calzone K.A., Stopfer J.E., Nathanson K.L., Weber B.L. Cancer risk estimates for BRCA1 mutation carriers identifi ed in a risk evaluation program. J Natl Cancer Inst. 2002; 94: 1365-72.
…
25. Zhang L., Kirchhoff T., Yee C.J., Offit K. A Rapid and Reliable Test for BRCA1 and BRCA2 Founder Mutation Analysis in Paraffin Tissue Using Pyrosequencing. The Journal of Molecular Diagnostics. 2009; 11(3): 176‑81.
Повний список літератури знаходиться в редакції.
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 3 (64) 2020 р.