


2 квітня, 2017
Профілі фармакокінетичних лікарських взаємодій інгібіторів протонної помпи: оновлені докази
Вступ
Порівняно з антагоністами Н2-рецепторів гістаміну, інгібітори протонної помпи (ІПП) забезпечують більш виражене і тривале пригнічення шлункової кислоти, а також кращий рівень виліковування при багатьох кислотозалежних захворюваннях шлунка. Завдяки цьому ІПП вважаються незамінними в лікуванні гастроезофагеальної рефлюксної хвороби (ГЕРХ), пептичної виразки шлунка або дванадцятипалої кишки, синдрому Золлінгера – Еллісона. ІПП також є ключовою складовою потрійної терапії (у комбінації з двома антибіотиками, такими як кларитроміцин, амоксицилін або метронідазол) для ерадикації Helicobacter pylori при пептичній виразці шлунка або дванадцятипалої кишки і може застосовуватись у профілактиці пептичних виразок, індукованих стресом та нестероїдними протизапальними засобами. Багато з цих захворювань у цілому потребують тривалого лікування, що підвищує ризик клінічно значимих лікарських взаємодій у пацієнтів (особливо в госпіталізованих хворих і пацієнтів літнього віку), які отримують ІПП й інші препарати.
Попередній огляд, опублікований у 2006 р., продемонстрував подібності й розбіжності серед ІПП стосовно вірогідності, значимості й механізмів взаємодії ліків (H. Blume et al., Pharmacokinetic drug interaction profiles of proton pump inhibitors. Drug Saf. 2006; 29 (9): 769-784). У ньому обговорювалося те, як ІПП шляхом підвищення рН можуть модифікувати вивільнення інших препаратів з їх лікарських форм у шлунку, а також яким чином ІПП впливають на абсорбцію й метаболізм ліків, взаємодіючи з аденозинтрифосфатзалежним Р-глікопротеїном або ферментною системою цитохрому Р450 (CYP). На момент проведення першого огляду найбільш вивченими були профілі взаємодії омепразолу й пантопразолу натрію (пантопразолу-Na).
Проаналізувавши великий об’єм доказів, автори дійшли висновку, що омепразол має значний потенціал лікарських взаємодій внаслідок його високої афінності до CYP2C19 і помірної афінності до CYP3A4, тоді як пантопразол-Na має нижчий потенціал взаємодій порівняно з омепразолом. Лансопразол і рабепразол також здавались такими, що мають більш слабкий потенціал взаємодій, ніж омепразол, проте це твердження ґрунтувалося лише на обмежених доказах. Значною мірою результати того огляду залишаються актуальними й сьогодні; утім, після 2006 р. було опубліковано декілька нових досліджень, присвячених лікарським взаємодіям ІПП. Отже, вашій увазі пропонується оновлення огляду 2006 р., яке разом з оригінальною статтею надає всебічний аналіз лікарських взаємодій, асоційованих із застосуванням цих препаратів.
Цей огляд ґрунтується на літературі, опублікованій з 1 січня 2007 до 31 грудня 2012 р. й ідентифікованій шляхом пошуку в базах даних MEDLINE (PUBMED) і EMBASE із використанням відповідних термінів і ключових слів. Пошук обмежували англомовними роботами і виключали коментарі, редакційні статті, листи, замітки та доклади з конференцій або огляди. Після об’єднання результатів PUBMED і EMBASE виключили дублікати; результати, які залишилися, розділили на статті щодо взаємодії ІПП з клопідогрелем і роботи, присвячені взаємодіям ІПП з іншими препаратами. Крім того, додаткові статті отримували шляхом пошуку вручну в списках літератури відповідних оглядів і наукових робіт. У цілому було знайдено 132 статті щодо взаємодії з клопідогрелем і 174 статті стосовно взаємодії з іншими препаратами. Два автори, ґрунтуючись на відповідному дизайні досліджень лікарських взаємодій, незалежно один від одного відібрали додаткові статті для включення в аналіз; будь-які невідповідності були обговорені й погоджені. Загалом у цей огляд увійшли 40 нових статей.
Механізми лікарських взаємодій ІПП
Зміна внутрішньошлункового рН
Взаємодії між ІПП та іншими препаратами, специфічні для всього класу ІПП, можуть виникати внаслідок ІПП-індукованого підвищення внутрішньошлункового рН, що може знижувати розчинну кількість діючої речовини інших препаратів, змінювати вивільнення препарату з лікарських форм, розчинність яких залежить від рН, або опосередковано впливати на біодоступність внаслідок зміни фармакокінетики проліків. Приклади фармакокінетики препаратів, яка залежить від внутрішньошлункового рН, докладно обговорювалися в огляді 2006 р. Останні включають знижену біодоступність кетоконазолу при його пероральному прийомі одночасно з прийомом омепразолу 60 мг, а також зменшену площу під фармакокінетичною кривою «концентрація-час» упродовж 24 год (AUC24) і знижену концентрацію в плазмі (Сmax) при пероральному прийомі ітраконазолу в формі капсул у разі призначення з омепразолом 40 мг.
Після публікації огляду 2006 р. з’явилися нові дані щодо взаємодії ІПП з мофетилу мікофенолатом. Призначення ІПП підвищує внутрішньошлунковий рН, що вповільнює гідроліз мофетилу мікофенолату, знижуючи максимальну експозицію і доступність мікофенолової кислоти, принаймні у ранніх точках часу. Порівняно з монотерапією мофетилу мікофенолатом, одночасне його призначення з пантопразолом-Na призводило до стабільно знижених плазмових концентрацій мікофенолової кислоти в пацієнтів з трансплантованим серцем і значного зниження загальної і максимальної експозиції в пацієнтів з аутоімунними захворюваннями. Це корелювало з підвищенням на 42% (р<0,01) площі активності інозинмонофосфатдегідрогенази. Проте одночасне призначення пантопразолу-Na і мікофенолату натрію з кишковорозчинним покриттям не супроводжувалося будь-якими значними змінами параметрів фармакокінетики в пацієнтів з трансплантованими легенями або серцем. Ці дані, отримані під час вивчення рівноважних концентрацій препаратів, підтверджують результати попереднього дослідження за участю здорових добровольців. У рівноважному стані пантопразол-Na (40 мг/добу) значно знижував загальну й максимальну експозицію мікофенолової кислоти після прийому мофетилу мікофенолату, проте не чинив значного ефекту в разі призначення мікофенолату натрію з кишковорозчинним покриттям. Інші фармакокінетичні параметри не змінювалися, що свідчить про малоймовірність взаємодії на ферментному рівні.
Інші взаємодії, які не обговорювались у попередньому огляді, включають зміни контакту самих ІПП зі шлунковим середовищем, що змінює експозицію ІПП. Цей ефект виникає переважно внаслідок нестабільності ІПП за низьких значень рН і обумовлює необхідність призначення ІПП у шлунково-резистентних лікарських формах. Відповідно, одночасний прийом прокінетика мосаприду призводить до підвищення загальної і максимальної експозиції після призначення рабепразолу приблизно на 50%, що пояснюється прискореним транспортом у тонку кишку. Ці результати доповнюють дані, отримані раніше для комбінації омепразолу і мосаприду, і свідчать про те, що така взаємодія буде спостерігатись і для всіх інших ІПП. Проте це пояснення не враховує того факту, що призначення у шлунково-резистентних формах означає відсутність контакту ІПП і шлункової кислоти, тобто можливі й інші, ще не виявлені фармакокінетичні взаємодії з мосапридом.
Класовий ефект з чіткими клінічними наслідками передбачається для багатьох інгібіторів протеаз, біодоступність яких може суттєво змінюватись у разі одночасного призначення з ІПП. Наприклад, загальна й максимальна експозиція однократної дози атазанавіру 400 мг знижувалася більш ніж на 90% за умови призначення з лансопразолом 60 мг. Вважається, що цей ефект зумовлений втратою розчинності атазанавіру за підвищених значень рН, оскільки CYP-опосередкована взаємодія є малоймовірною для цієї комбінації препаратів. Для інших комбінацій взаємодії можуть бути більш складними. Експозиція нелфінавіру, розчинність якого подібним чином залежить від рН, у рівноважному стані (після 4 днів прийому в дозі 1250 мг 2 р/добу) знижувалася приблизно на 35% у разі одночасного лікування омепразолом 40 мг 1 р/добу протягом 4 днів, проте кінцеві виведення і кліренс залишалися незмінними. Утім, нелфінавір метаболізується CYP2C19, інгібування якого омепразолом може протидіяти втраті експозиції внаслідок зниження розчинності. Це також пояснює зниження метаболічного співвідношення головного метаболіту й нелфінавіру.
На відміну від атазанавіру й нелфінавіру, загальна й максимальна експозиція однократної дози ралтегравіру 400 мг у разі призначення з омепразолом (20 мг 1 р/добу 4 дні) підвищується у 3 і 4 рази відповідно. Враховуючи шлях метаболізму ралтегравіру, взаємодії на ферментному рівні є малоймовірними. Натомість ралтегравір має суттєво підвищену розчинність за високих значень рН і є субстратом для Р-глікопротеїну, який принаймні помірно пригнічується омепразолом, і обидва ці ефекти можуть бути синергічними. Отже, слід враховувати не лише потенційні класові ефекти ІПП, а й індивідуальні взаємодії кожного препарату.
Ситуація з інгібіторами протеаз ускладнюється ще більше в разі супутнього використання ритонавіру, що відбувається досить часто. Ритонавір, який краще розчиняється за низьких значень рН, посилює активність інших інгібіторів протеаз шляхом пригнічення CYP3A4, метаболізується CYP3A4 (подібно до ІПП) і є субстратом й інгібітором Р-глікопротеїну.
Загальна і максимальна експозиція лопінавіру й ритонавіру в рівноважному стані підвищувалася приблизно на 25% при одночасному застосуванні з омепразолом без суттєвих змін елімінації. Цей результат пояснюється підвищенням інгібування Р-глікопротеїну омепразолом і, як наслідок, більш сильним інгібіторним ефектом ритонавіру на CYP3A4. В іншому дослідженні прийом інгібітора протеаз і омепразолу з інтервалом 2 год значно послабив цей ефект; підвищення загальної і максимальної експозиції ритонавіру після розділення дозувань зменшилося з 14 до 3% і з 16 до 8% відповідно. Проте експозиція одночасно призначеного саквінавіру залишилася підвищеною на 50-70%, отже, вона практично не залежала від зміни експозиції ритонавіру (пояснюється наявністю іншого, більш системного ефекту). Ці дані підтверджуються результатами дослідження, в якому підвищення експозиції ритонавіру було мізерним, проте експозиція саквінавіру збільшилася приблизно на 80% при одночасному прийомі омепразолу.
Поєднаний вплив багатьох факторів був продемонстрований у дослідженні з однократним прийомом індинавіру 800 мг, експозиція якого знижувалася на 35 і 45% при постійному лікуванні омепразолом 20 і 40 мг, проте підвищувалася на 55% у разі додавання однократної дози ритонавіру до високої дози омепразолу.
Взаємодії з аденозинтрифосфатзалежним ефлюксним переносником Р-глікопротеїном
Після публікації попереднього огляду нові дослідження стосовно ІПП і Р-глікопротеїнової транспортної системи не проводилися. Згідно з наявними даними, омепразол, лансопразол і пантопразол-Na, які всі є субстратами Р-глікопротеїну, інгібували опосередкований Р-глікопротеїном ефлюкс дигоксину в клітинній моделі Caco‑2. Езомепразол і рабепразол у цьому дослідженні не вивчалися.
Система цитохрому Р450
Взаємодії з кишковими й печінковими ферментами системи CYP докладно обговорювалися в огляді 2006 р. і не будуть згадуватися у цій статті, однак важливо нагадати, що ІПП метаболізуються переважно в печінці ферментами CYP2C19 і CYP3A4.
Після публікації попереднього огляду багато метааналізів та оглядів були присвячені лікарським взаємодіям між певними ІПП і клопідогрелем. Ці взаємодії, імовірно, опосередковуються CYP2C19 і мають надзвичайну клінічну значимість. Нещодавні ретроспективні дослідження вказували на послаблення позитивних ефектів клопідогрелю при одночасному лікуванні ІПП, проте стратифікація аналізу свідчить про те, що цього не відбувається в пацієнтів, які приймають пантопразол-Na, порівняно з хворими, які використовують омепразол. У багатьох дослідженнях було продемонстровано, що в рівноважному стані омепразол значно підвищує загальну експозицію клопідогрелю і знижує експозицію його активного метаболіту. Ці ефекти зберігаються навіть у разі відокремленого призначення препаратів з інтервалом 12 год або за умови використання подвійних доз клопідогрелю. Однак ці зміни явно зменшувалися після заміни омепразолу пантопразолом натрію.
Вищенаведені дані узгоджуються з результатами дослідження, в якому було встановлено, що клопідогрель має активуватися CYP2C19-ферментом, який інгібується омепразолом, а не пантопразолом-Na. Також було визначено, що при одночасному призначенні омепразолу або езомепразолу експозиція активного метаболіту клопідогрелю і пригнічення функції тромбоцитів значно зменшуються. Значимість CYP2C19 підкреслюється результатами дослідження, в якому спостерігався лише незначний ефект при одночасному призначенні лансопразолу і прасугрелу (останній переважно активується іншими ізоферментами CYP).
Ситуація з лансопразолом видається більш складною, проте, на відміну від рабепразолу, щодо цього ІПП наявна хоча б обмежена інформація, у тому числі дані фармакокінетики. Одночасне призначення лансопразолу і клопідогрелю не впливало на утворення неактивного карбоксильного метаболіту клопідогрелю. Проте фармакодинамічний ефект значно знижувався в пацієнтів з попередньою високою відповіддю на клопідогрель, вірогідно, внаслідок інгібування активації клопідогрелю ферментом CYP2С19, який не бере участь в утворенні карбоксильного метаболіту (останній формується переважно під дією естераз). Проте аналіз усіх пацієнтів у цьому дослідженні показав лише тенденцію до зниженої ефективності клопідогрелю. Це збігається з результатами іншого дослідження, в якому було продемонстровано, що лансопразол і декслансопразол не спричиняють значного впливу на експозицію активного метаболіту або фармакокінетику клопідогрелю.
Таким чином, взаємодія між клопідогрелем та ІПП достовірно має місце для омепразолу й езомепразолу, тоді як щодо рабепразолу наявні дані обмежені. Декслансопразол, лансопразол і пантопразол-Na мають менший вплив на антитромбоцитарну активність клопідогрелю порівняно з омепразолом або езомепразолом, що також зазначено в інструкції до препарату Плавікс.
Профілі взаємодії ІПП
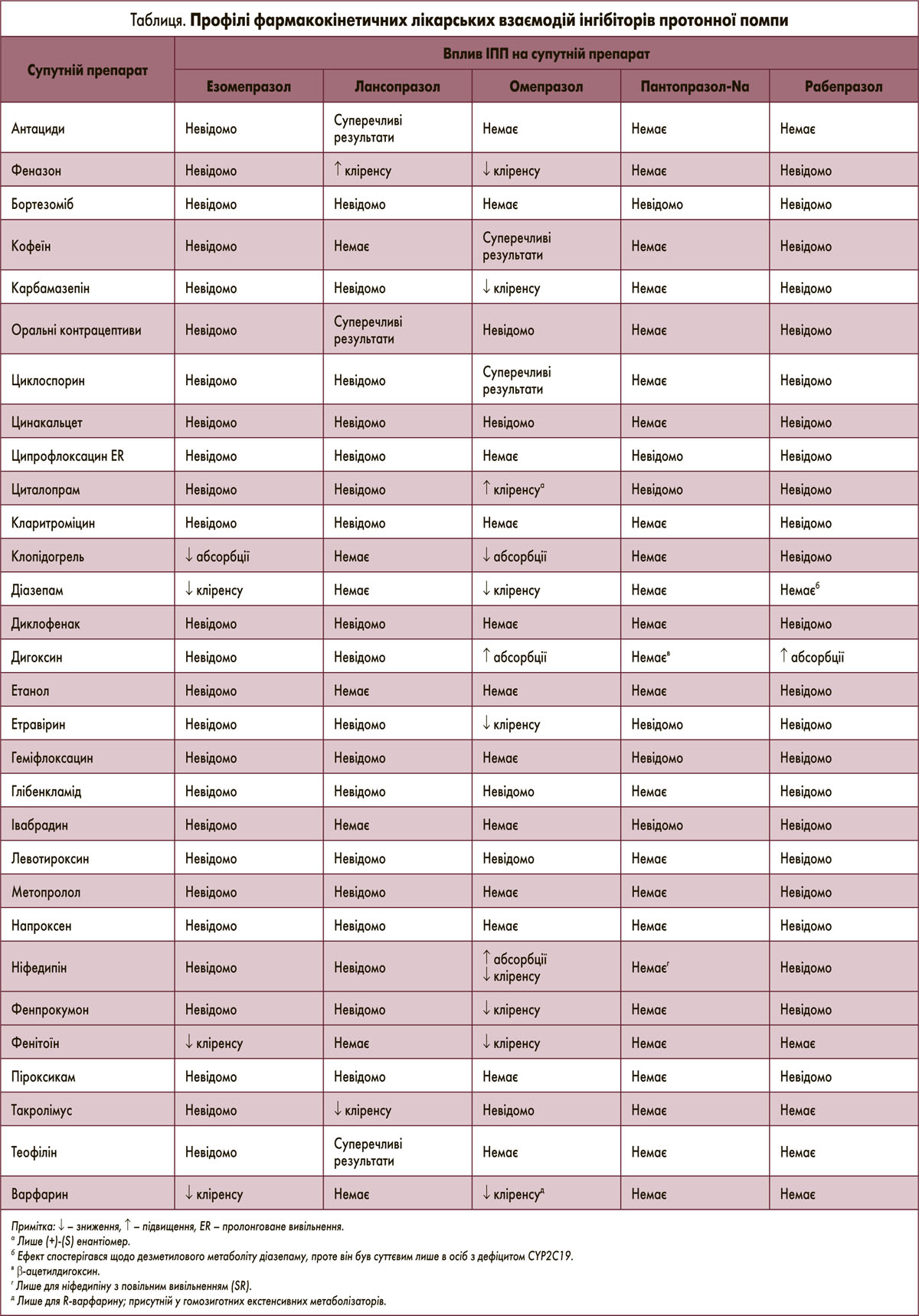
Профілі взаємодії омепразолу й пантопразолу-Na є добре вивченими, тоді як дані стосовно езомепразолу, лансопразолу й рабепразолу визначені меншою мірою. Основні результати цих досліджень наведені в таблиці; остання також містить нові дані щодо взаємодії ІПП з такими препаратами, як бортезоміб, ципрофлоксацин подовженого вивільнення, циталопрам, кларитроміцин, клопідогрель, етравірин, геміфлоксацин та івабрадин.
Ретроспективне дослідження типу «випадок-контроль», проведене після 2006 р., виявило підвищену залишкову агрегацію й активацію тромбоцитів у пацієнтів з ішемічною хворобою серця, які одночасно отримували ацетилсаліцилову кислоту (75 мг на добу, без кишковорозчинної оболонки) та ІПП. У проспективних дослідженнях одночасне призначення ацетилсаліцилової кислоти з кишковорозчинною оболонкою та пантопразолу-Na супроводжувалося зниженням агрегації тромбоцитів, а призначення лансопразолу суттєво не впливало на активність тромбоцитів або рівні саліцилатів у крові. Отже, результати цих проспективних досліджень дії ІПП при одночасному прийомі у близько 70% пацієнтів дослідження «випадок-контроль» не підтверджують порушення дії ацетилсаліцилової кислоти. Вплив омепразолу й езомепразолу на тромбоцитарну функцію не може бути виключений; у випадках одночасного призначення цих ІПП з ацетилсаліциловою кислотою рекомендується моніторинг ефективності лікування.
Окремі клінічні спостереження (за даними літератури й системи повідомлення про небажані реакції FDA США) і популяційні фармакокінетичні дослідження вказують на те, що одночасне призначення ІПП і метотрексату може підвищувати і подовжувати сироваткові рівні метотрексату та/або його метаболіту гідроксиметотрексату, хоча механізм цієї взаємодії остаточно не з’ясований.
Омепразол
У попередньому огляді повідомлялося про взаємодії омепразолу з діазепамом, прогуанілом і антидепресантом моклобемідом (в активних метаболізаторів) за рахунок конкурентного інгібування CYP2C19. Омепразол-індуковане конкурентне інгібування CYP2C19 також може змінювати метаболізм фенітоїну і варфарину. Нещодавно було встановлено, що інгібування CYP2C19 омепразолом знижує кліренс (+)-(S)-циталопраму на 50% з відповідним підвищенням плазмової концентрації на 120% у здорових добровольців, а також підвищує загальну експозицію етравірину на 41% після прийому однократної дози етравірину 100 мг і багатократного прийому омепразолу (у разі багатократного прийому ранітидину цей ефект не спостерігався).
Також вивчався вплив омепразолу на фармакокінетику антацидів, борезомібу, ципрофлоксацину сповільненого вивільнення, геміфлоксацину, ніфедипіну, метопрололу, нестероїдних протизапальних засобів, заліза і теофіліну, проте клінічно значимих взаємодій не було виявлено. Систематичні клінічні дослідження показали суперечливі результати щодо взаємодії між омепразолом і циклоспорином: концентрації циклоспорину підвищувались у пацієнтів із трансплантованим серцем, проте не змінювались у пацієнтів із трансплантованою ниркою.
Препарати з високою афінністю до CYP3A4 (наприклад, кетоконазол, флуконазол, кларитроміцин, моклобемід) можуть впливати на біодоступність омепразолу шляхом підвищення його сироваткової концентрації, проте це має клінічну значимість лише в пацієнтів з дефіцитом CYP2C19, в яких метаболізм омепразолу відбувається за рахунок CYP3A4.
Кінетика омепразолу також залежить від CYP2C19. Знижені плазмові концентрації омепразолу і сульфону омепразолу спостерігалися після призначення гінкго білоба і звіробою. Метаболізм омепразолу знижувався після призначення флувоксаміну (лише в активних метаболізаторів), і AUC омепразолу збільшувалася після використання комбінованого орального контрацептиву, який містив етинілестрадіол.
Таким чином, омепразол має численні лікарські взаємодії, хоча не всі вони є клінічно значимими. Велика кількість повідомлень про взаємодії також може пояснюватися тим, що омепразол присутній на фармринку довше порівняно з іншими ІПП (з 1989 р.).
Езомепразол
Нових даних щодо CYP-опосередкованих взаємодій езомепразолу немає. Огляд 2006 р. дійшов висновку, що потенціал взаємодій езомепразолу й рацемічного омепразолу суттєво не відрізняється. Езомепразол, вірогідно, не взаємодіє з препаратами, які метаболізуються переважно CYP1A2, CYP2A6, CYP2C9, CYP2D6 або CYP2E1, проте така взаємодія відбувається з препаратами, що метаболізуються CYP2C19. Це було продемонстровано в дослідженнях з фенітоїном і R-варфарином, хоча ці взаємодії не досягли клінічної значимості. Крім того, багаторазовий прийом езомепразолу підвищував концентрації діазепаму й уповільнював його елімінацію, чого не відбувалось у разі застосування пантопразолу-Na. Ці ефекти езомепразолу клінічно проявлялися порушеннями рухової координації і пильності.
Пантопразол
Останнім часом було доведено, що пантопразол-Na не має значимих взаємодій із клопідогрелем. Автори попереднього огляду дійшли висновку, що численні дослідження за участю здорових добровольців і пацієнтів із різними захворюваннями продемонстрували низький потенціал взаємодії пантопразолу-Na з іншими препаратами. Так, були відсутні значимі метаболічні взаємодії при комбінуванні пантопразолу-Na з антацидами, фенозоном (антипірином), кофеїном, карбамазепіном, цинакальцетом, кларитроміцином, циклоспорином, клопідогрелем, діазепамом, диклофенаком, β-ацетилдигоксином, етанолом, глібенкламідом, левотироксином, метопрололом, напроксеном, ніфедипіном тривалого вивільнення, оральними контрацептивами, фенпрокумоном, фенітоїном, піроксикамом, такролімусом, теофіліном або варфарином. Було знайдено клінічно незначну взаємодію між пантопразолом-Na 40 мг та цизапридом 20 мг.
Пантопразол-магній (пантопразол-Mg) – поліпшена форма пантопразолу, яка була розроблена пізніше огляду 2006 р. У пантопразолі-Mg до активної речовини замість натрієвої солі додається магнієва сіль. Оскільки пантопразол-Na і пантопразол-Mg є різними солями тієї самої молекули, їх профілі лікарських взаємодій, вірогідно, є подібними.
Лансопразол
Після огляду 2006 р. нові дослідження щодо CYP-опосередкованих взаємодій лансопразолу не проводилися. Як повідомлялося раніше, лансопразол не має клінічно значимих взаємодій з феназоном, діазепамом, івабрадином, магалдратом, оральними контрацептивами, фенітоїном, преднізолоном, пропранололом і варфарином. Підвищення біодоступності теофіліну після призначення лансопразолу не вважається клінічно значимим, і підвищений кліренс теофіліну на тлі лікування лансопразолом спостерігається не в усіх випадках. Лансопразол знижував кліренс такролімусу при пероральному прийомі, значно підвищуючи концентрації останнього в крові, особливо в пацієнтів з мутантними алелями CYP2C19. Інгібітор CYP2C19 флувоксамін спричиняє значний вплив на метаболізм лансопразолу (підвищує концентрацію в плазмі) в активних метаболізаторів щодо CYP2C19, але не в слабких метаболізаторів.
Нещодавно на фармринку з’явився декслансопразол – активний енантіомер лансопразолу. Ця сполука доступна в інноваційній лікарській формі двофазного вивільнення, що дозволяє подовжити профіль плазмової концентрації в часі після прийому 1 раз на добу. З огляду на те що механізмом дії ІПП є незворотне інгібування протонної помпи, клінічні переваги такого біофармацевтичного профілю слід докладно дослідити в клінічній практиці. Взаємодії декслансопразолу подвійного вивільнення вивчались щодо діазепаму, фенітоїну, теофіліну й варфарину як «зондових» препаратів (стосовно CYP2C19, CYP2C9, CYP1A2 і CYP3A); впливу на фармакокінетику цих сполук виявлено не було.
Загалом профілі взаємодії лансопразолу й декслансопразолу вивчені меншою мірою, ніж омепразолу й пантопразолу-Na. Утім, жоден із цих препаратів, вірогідно, не асоціюється з клінічно значимими лікарськими взаємодіями.
Рабепразол
Після огляду 2006 р. інформація щодо лікарських взаємодій рабепразолу не змінилася. Взаємодії цього ІПП є менш вивченими, ніж омепразолу й пантопразолу-Na (табл.). Більшість досліджень повідомляли про взаємодії, властиві всім ІПП внаслідок зміни внутрішньошлункового рН (наприклад, взаємодії з дигоксином й кетоконазолом). Значимі CYP-опосередковані лікарські взаємодії з рабепразолом у цілому є малоймовірними, оскільки він має низьку афінність до багатьох ізоферментів CYP. Проте для підтвердження цього потрібні відповідні дослідження. В огляді 2006 р. зазначалася відсутність даних про метаболічні взаємодії рабепразолу з теофіліном, варфарином, фенітоїном, такролімусом і антацидами. Вплив рабепразолу на фармакокінетику десметилового метаболіту діазепаму був значимим лише у слабких метаболізаторів стосовно S-мефенітоїн‑4’гідроксилювання (наприклад, у пацієнтів з дефіцитом CYP2C19).
Інгібітор CYP2C19 флувоксамін суттєво впливає на метаболізм рабепразолу в гомо- і гетерозиготних активних метаболізаторів щодо CYP2C19, підвищуючи AUC(0,∞) і час напіввиведення рабепразолу і його тіоефіру. У слабких метаболізаторів (*2/*2) подібний ефект не спостерігається.
Висновки
Огляд відповідних досліджень, опублікованих після 2006 р., виявив додаткові взаємодії ІПП, опосередковані внутрішньошлунковим рН (наприклад, з мофетилу мікофенолатом), нестабільністю ІПП за низьких значень рН, а також зміненою фармакокінетикою багатьох інгібіторів протеаз (у тому числі атазанавіру, нелфінавіру, ралтегравіру, ритонавіру й індинавіру). Нові дані щодо CYP-опосередкованих взаємодій стосуються передусім декслансопразолу та взаємодії ІПП з клопідогрелем. Серед клінічно значимих взаємодій заслуговує на увагу взаємодія між клопідогрелем та омепразолом або езомепразолом внаслідок механізму, не властивого всьому класу ІПП. Крім того, до проведення додаткових досліджень не можна виключити вплив омепразолу й езомепразолу на агрегацію тромбоцитів у разі одночасного призначення з ацетилсаліциловою кислотою. Одночасне призначення ІПП і метотрексату може впливати на фармакокінетику останнього, хоча механізм цієї взаємодії вивчений недостатньо.
У цілому висновки огляду 2006 р. зберігають свою значимість. Вбачається, що лансопразол, пантопразол-Na і рабепразол асоціюються з нижчою частотою лікарських взаємодій порівняно з омепразолом і езомепразолом завдяки більш низькій афінності до окремих ізоферментів CYP або залученню додаткових процесів елімінації. Утім, слід наголосити, що лише профіль взаємодії пантопразолу-Na є добре вивченим.
З урахуванням незначних відмінностей між ІПП стосовно клінічної ефективності (за використання в еквівалентних дозах) розбіжності у профілях лікарських взаємодій стають важливим фактором при виборі препарату, особливо в пацієнтів, які вже приймають багато супутніх лікарських засобів (хворі літнього віку) або отримують препарати з вузьким терапевтичним вікном. ІПП з добре доведеним низьким ризиком лікарської взаємодії будуть найкращим вибором для таких пацієнтів.
Список літератури знаходиться в редакції.
Wedemeyer R.-S., Blume H. Pharmacokinetic Drug Interaction Profiles of Proton Pump Inhibitors: An Update. Drug Saf. 2014; 37: 201-211.
Переклав з англ. Олексій Терещенко
UA/(PPIF)/0117/0004
За підтримки ТОВ «Такеда України»