


19 травня, 2017
Діабетична хвороба нирок
 М.В. Власенко
М.В. Власенко
Історія відкриття патологічних змін у нирках при цукровому діабеті (ЦД) налічує понад століття. У 1840 році в паризькому госпіталі С. Bernard, виконуючи патоморфологічне дослідження пацієнта з уперше виявленим ЦД, зафіксував виражене збільшення розмірів нирки [1, 2]. Термінальну стадію діабетичної нефропатії (ДН) уперше описали ще в 1936 році P. Kimmеlstiel і C. Wilson. Відтак було висунуто гіпотезу, що нефропатія зумовлена гіперглікемією [3]. За час, що минув відтоді, було досягнуто певного прогресу й розуміння природи зазначеного ускладнення, механізмів його розвитку та методів впливу на них. Утім, незважаючи на досягнуті успіхи, про вирішення цієї проблеми наразі не йдеться.
На сьогодні ДН є найбільш серйозним ускладненням ЦД, посідає високі позиції у структурі компонента дорослих хворих, які потребують проведення гемо- або перитонеального діалізу [4]. Тривалий час нефропатію діагностували у стадії, коли розвивалися симптоми нефротичного синдрому й формувалася хронічна ниркова недостатність (ХНН). Із 80-х років минулого століття виникла можливість діагностувати клінічні стадії ДН і проводити її профілактику [5]. Частота появи ДН безпосередньо залежить від тривалості ЦД. Цей зв’язок встановлений у хворих на ЦД 1 типу. В перші 5 років від початку ЦД 1 типу, який має відносно точну дату свого початку, оскільки пов’язаний з інсуліновою недостатністю, ДН не розвивається. Хоча в окремих роботах (R.P. Hamman, 1997) зазначалося, що у хворих із ЦД 1 типу з тривалістю захворювання до 5 років у 18% спостерігалася альбумінурія. Більш виражена стадія ДН – стадія протеїнурії – рідко розвивається упродовж 5 років від початку ЦД 1 типу.
За даними клініки Stena (м. Копенгаген), ДН у хворих на ЦД 1 типу з тривалістю діабету до 10 років становить 5-6%, при тривалості захворювання до 20 років – 25-30%, до 30 років – 35-40% [6]. У разі тривалості захворювання на ЦД понад 30-35 років, якщо ДН не розвинулася, ймовірність її виникнення становить 1% нових випадків на рік. Це свідчить про можливість генетичної захищеності цих пацієнтів від патологічного впливу метаболічних факторів на нирки. Встановлено, що частота ДН при ЦД 1 типу залежить від того, у якому віці почалося захворювання. Так, частота виявлення ДН досягала 44-45% в осіб із розвитком ЦД у пубертатному віці 15-20 років. Якщо ЦД розпочинався, коли пацієнтові виповнилося 20 років, то ДН траплялася у 30-35% випадків. Припускають, що вікові відмінності у прогресуванні ДН пов’язані з патологічним впливом метаболічних порушень у цей період, зумовлених гормональною перебудовою організму (активна секреція гормона росту й статевих гормонів). Поширеність ДН при ЦД 2 типу менше вивчено, оскільки встановлення точного часу початку захворювання ускладнено. ЦД 2 типу розвивається в осіб немолодого віку, тож при цьому симптоми захворювання можуть нашаровуватися на прояви інших супутніх захворювань, які з’явилися до початку ЦД: гіпертонічна хвороба та інша серцево-судинна патологія, гломерулонефрит, пієлонефрит, сечокам’яна хвороба. Більш того, альбумінурія в 15-40% випадків виявляється при першому виявленні ЦД 2 типу або навіть передує встановленому діагнозу [6]. При першому виявленні ЦД 2 типу протеїнурія зустрічається у 7-10% хворих [6]. Однак при ЦД 2 типу простежується так само, як і при ЦД 1 типу, залежність частоти розвитку ДН від тривалості захворювання. ДН спостерігається у 7-10% хворих із ЦД 2 типу тривалістю не більше 5 років, 20-35% – у пацієнтів із ЦД 2 типу зі стажем 20-25 років і 50-57% – при більш тривалих термінах захворювання [7]. Незважаючи на подібну частоту розвитку ДН у хворих на ЦД 1 і 2 типу, смертельний наслідок від термінальної ниркової недостатності в пацієнтів із ЦД 2 типу настає рідше, ніж у хворих на ЦД 1 типу (5-10% випадків при ЦД 2 типу проти 45% при ЦД 1 типу). Це пов’язано з тим, що у хворих із ЦД 2 типу на тлі нефропатії спостерігається весь клінічний спектр серцево-судинної патології, тож смерть цих хворих насамперед настає від серцево-судинної катастрофи. Необхідно зазначити, що при ЦД 2 типу (30% хворих) і при ЦД 1 типу (8-12% хворих) виявлена протеїнурія може свідчити, що ураження нирок не пов’язане із ЦД, тому необхідно проводити диференційний діагноз.
Стереотипне уявлення про ДН може маскувати різні захворювання нирок, зокрема діабетичний громерулосклероз, ішемічну нефропатію, інфекцію сечових шляхів, гіпертонічний нефросклероз, інтерстиціальний нефрит, гломерулонефрит тощо. Це пов’язано з особливостями метаболічних порушень, властивих ЦД. Для цього захворювання характерні мікро- і макроангіопатії, схильність до інфекційних ускладнень, підвищений ризик серцево-судинних захворювань.
Опубліковано результати обстеження пацієнтів із ЦД 2 типу у віці 40 років і старше. ХНН встановлено у 13% хворих, серед них альбумінурію зафіксовано лише у 28%, протеїнурію – у 19%, діабетичну ретинопатію (ДР) – у 28%.
У 30% хворих із ЦД 2 типу з ХНН була відсутня як ретинопатія, так і альбумінурія [8].
Це дало змогу зробити висновок, що патологія нирок у хворих на ЦД 2 типу часто зумовлена захворюваннями паренхіми нирок, які відрізняються від діабетичного гломерулосклерозу, й рекомендувати обов’язкове визначення швидкості клубочкової фільтрації (ШКФ) як доповнення до моніторингу екскреції альбуміну та оцінки змін судин очного дна. Взаємозв’язок ДР і ДН у хворих на ЦД 2 типу – особлива тема. ДР встановлюють у майже 90% хворих на ЦД 1 типу з протеїнурією і тільки у 50% хворих із ЦД 2 типу [8]. Це може відображати наявність у нирках патологічних процесів, які не залежать від ефектів гіперглікемії, а також мають різний термін розвитку і диференційовану відповідь на дію факторів, які впливають на зниження ниркової функції. Більш того, у разі ізольованої ДН хворі мають кращий нирковий і кардіальний прогноз незалежно від контролю рівня артеріального тиску (АТ) і метаболічних показників.
Наявність ДР як одного з базових показників асоціюється із більш значними ризиками зростання рівня креатиніну в сироватці крові, кінцевої стадії ХНН, летальності [8]. На підставі аналізу факторів ризику зниження ШКФ, збільшення креатиніну в крові та смертності було встановлено значущість таких факторів, як рівень альбуміну, систолічний рівень АТ, рівень глікованого гемоглобіну крові (HbAlc), паління та вік пацієнта [8].
Протеїнурію розглядають як надзвичайно важливий негемодинамічний предиктор прогресування ДН. Редукція протеїнурії до менш ніж 1 г/добу є такою ж важливою, як і мета досягнення цільових показників глікемії, АТ, ліпідів сироватки крові для запобігання прогресуванню ДН.
У країнах Європи чітко встановлено подвоєння кількості пацієнтів, які хворіють на ЦД, із термінальною стадією ниркової недостатності (за 9-річний період спостереження – із 12,7 до 23,6 на 1 млн населення) [9]. Навіть у країнах із відносно низькою захворюваністю на ЦД – Австралії та Новій Зеландії – відзначено майже двократне зростання кількості хворих на термінальну ниркову недостатність за рахунок пацієнтів із ЦД 2 типу [10]. Менш урахованою і дослідженою залишається група хворих на ЦД із помірною нирковою недостатністю, що ускладнює прогнозування динаміки поширеності термінальної ниркової недостатності й потреби в замісній нирковій терапії (ЗНТ). Діабетологія і нефрологія – доволі витратні галузі охорони здоров’я. Економічні витрати, спрямовані на забезпечення ЗНТ хворим ЦД, продовжують зростати.
ДН – специфічне ураження нирок при ЦД, що супроводжується формуванням вузликового або дифузного гломерулосклерозу, термінальна стадія якого характеризується розвитком ХНН.
Сучасні уявлення про патогенез ДН
Наразі не існує єдиного уявлення про механізми розвитку ДН. Згідно з результатами численних досліджень сьогодні основну увагу приділяють таким чинникам:
- порушення внутрішньониркової гемодинаміки у вигляді клубочкової гіпертензії, яка відіграє визначальну роль на ранніх стадіях ураження нирок при ЦД;
- пряма токсична дія альбуміну на епітеліоцити проксимальних канальців із подальшою індукцією процесів запалення та фіброзу в тубулоінтерстиціальному просторі;
- формування ендотеліальної дисфункції капілярів ниркових клубочків. Ключовим моментом у розумінні патогенезу ДН було відкриття В.М. Вгеппег (1996) феномена гіперфільтрації і внутрішньоклубочкової гіпертензії. Цей механізм запускається хронічною гіперглікемією, викликаючи спочатку функціональні, а згодом структурні зміни в нирках. Внутрішньоклубочкова гіпертензія розвивається внаслідок дилатації приносної артеріоли клубочка під дією надлишкової кількості глюкози, а також глюкагону, простацикліну, оксиду азоту (NO). Тривала дія потужного гідравлічного преса ініціює механічне подразнення прилеглих структур клубочка, що сприяє гіперпродукції колагену і його накопиченню в ділянці мезангіуму, початком склеротичних процесів, порушенням архітектоніки і проникності базальної мембрани клубочка. Іще одним важливим відкриттям було визначення надвисокої активності локальної ренін-ангіотензин-альдостеронової системи при ЦД. Встановлено, що локальна ниркова концентрація ангіотензину II (АГ ІІ) в 1000 разів перевищує його вміст у плазмі. Механізми його патогенної дії при ЦД зумовлені не лише його потужною вазоконстрикторною дією, а й проліферативною, прооксидантною і протромбогенною активністю. У нирках АГ ІІ викликає внутрішньоклубочкову гіпертензію, сприяє склерозуванню і фіброзуванню ниркової тканини опосередковано через викид цитокінів і факторів росту. Гіперглікемії належить провідна роль у розвитку мікро- та макросудинних ускладнень.
Гіперглікемія індукує неферментне глікірування білків, окислювальний стрес, активацію протеїну С, мітоген-активуючого протеїну, дію чинників зростання, вазоактивних факторів, цитокінів, що спричиняють ушкодження нирок на рівні клітини. Це призводить до розвитку ниркової гіпертрофії та акумуляції екстрацелюлярного матриксу, що передують таким незворотним змінам, як гломерулосклероз і тубулоінтерстиціальний фіброз. Підтверджують роль гіперглікемії у розвитку ДН результати великого дослідження DССТ (The Diabetes Control and Complications Trial Research Group), в ході якого було виявлено можливість запобігти розвитку ДН у хворих на ЦД 1 типу при ідеальній компенсації вуглеводного обміну і у хворих на ЦД 2 типу – дослідження UKPDS (UK Prospective Diabetes Study Group) і АDVANCE (Action in Diabetes and Vascular Disease).
Прогресуванню ДН також сприяють обмінні порушення у вигляді дисліпопротеїнемії, як завдяки здатності ліпопротеїдів низької і дуже низької щільності індукувати процеси фіброгенезу, так і гіперурикемії. Механізми розвитку тубулоінтерстиціального компонента запалення і фіброзу є універсальними як для ДН, так і для хронічних недіабетичних захворювань нирок. Значення протеїнурії як маркера прогресування уражень нирок багато в чому зумовлене механізмами токсичної дії окремих компонентів білкового ультрафільтрату на епітеліоцити проксимальних канальців та інші структури ниркового тубулоінтерстицію. Відомо, що білки плазми індукують процеси тубулоінтерстиціального запалення і фіброзу, які називають протеїнуричним ремоделюванням тубулоінтерстицію, вираженість якого є одним з основних факторів, що визначають швидкість прогресування ниркової недостатності при хронічних нефропатіях. Дослідження RENAAL (Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan), в яке було залучено пацієнтів із ЦД 2 типу і ДН, продемонструвало протеїнурію як найбільш значущий фактор ризику кардіоваскулярних подій і прогресування ДН незалежно від рівня АТ. Артеріальна гіпертензія (АГ) відіграє ключову роль у розвитку та прогресуванні ДН, так само як і в розвитку макроваскулярної патології. При прогресуванні ДН роль метаболічних факторів зменшується, натомість зростає роль гемодинамічних факторів (АГ, внутрішньоклубочкової гіпертензії).
Запобігти розвитку і прогресуванню судинних ускладнень (у тому числі ДН) можна тільки за підтримки АТ на рівні не більш як 130/80 мм рт. ст. Більш жорсткий контроль АТ у хворих із нирковою патологією може призвести до зростання ризику кардіоваскулярних подій через гіпоперфузію інших органів-мішенів. Велику увагу приділяють дослідженню розвитку анемії при прогресуванні ДН. Встановлено, що зниження рівня Hb у хворих із ЦД більш виражене порівняно з хворими з недіабетичними причинами хронічної хвороби нирок (ХХН) при зіставних величинах ШКФ. Рекомендації NKF K/DOQI (National Kidney Foundation) визначили цільові значення Hb у хворих ХХН, які отримують терапію еритропоетином у межах 110-120 г/л, але не вище 130 г/л через ризик більш частого виникнення мозкових інсультів. Довгий час тривали дебати про специфічність прояву ДН при ЦД 1 і 2 типу. Наразі існують доволі переконливі свідчення, що базові патофізіологічні механізми, які ведуть до розвитку й прогресування ДН, однакові при обох типах діабету. Однак при СД 2 типу, а також при метаболічному синдромі спостерігаються додаткові фактори ушкодження нирок, такі як ожиріння, гіперурикемія, дисліпідемія тощо. Деякі експерти вважають, що існує генетична схильність до діабетичного ураження нирок, хоча достовірних генетичних маркерів наразі не виявлено; крім того, не підтверджено теорію імунних факторів, тобто наявності антитіл до базальної мембрани клубочків та інсуліну.
Сучасне формулювання діагнозу
Клініко-морфологічна класифікація ДН. У зв’язку із зростанням кількості пацієнтів із нирковою недостатністю, яка розвивається унаслідок різних нозологічних форм, у 2002 році Національний фонд США запропонував об’єднати захворювання нирок із відомою причиною, з патологією, яка супроводжується зниженням ШКФ менше 60 мл/хв/1,73 м2 протягом трьох місяців і більше навіть за відсутності лабораторно-інструментальних ознак ураження нирок, незалежно від діагнозу в термін «хронічна хвороба нирок» (K/DOQ1-2002) [11]. Рівень ШКФ на сьогодні визнано кращим методом оцінки функції нирок. Нормальний рівень ШКФ співвідноситься з віком, статтю, площею поверхні тіла. Рівень ШКФ<60 мл/хв/1,75 м2 – порогова величина, що позначає втрату 50% фільтраційної здатності нормальної нирки. В міжнародній нефрологічній практиці частіше використовують класифікацію C.Е. Mogensen, основною перевагою якої є її орієнтація на виявлення ранніх стадій ДН [12]. З огляду на необхідність більш ранньої діагностики ДН, виникає запитання, на якій стадії лікар має можливість верифікувати ДН й розпочинати терапевтичні заходи щодо стримування прогресування ДН. Відповідно до класифікації C.Е. Mogensen розвиток ДН характеризується певними стадіями (табл. 1).
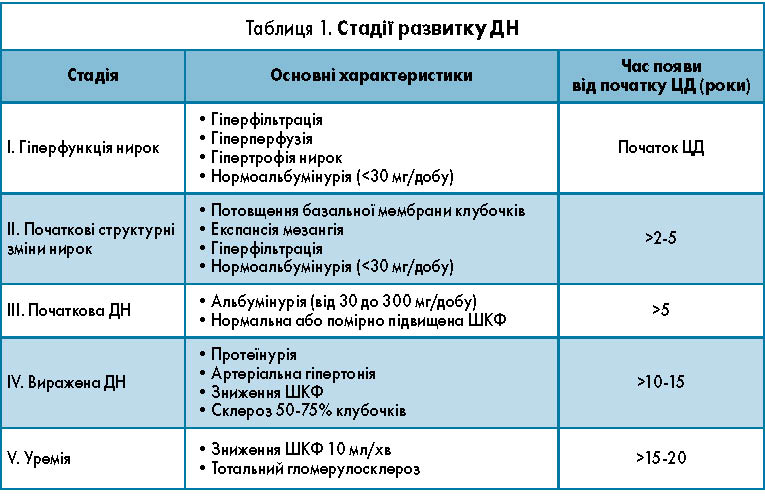
І стадія гіпертрофії нирок характеризується появою перших функціональних порушень у роботі нирок, які клінічно не проявляються. Екскреція білка із сечею – в нормі, АТ – в межах норми, набрякового синдрому немає. Однак ранні функціональні порушення нирок при ЦД виявляються в дебюті захворювання. Ці зміни характеризуються розвитком так званого процесу «гіперфільтрації», тобто підвищенням ШКФ вище нормальних значень (>140 мл/хв) і збільшенням ниркового кровотоку «гіперперфузії нирок». Встановлено, що у хворих із ЦД 1 типу в перші місяці після появи гіперглікемії ШКФ підвищується на 20-40% порівняно з нормою; це реакція нирок на стійку гіперглікемію. Однак така пряма залежність між глікемією і ШКФ зберігається при помірно високих значеннях цукру крові (до 13-14 ммоль/л); при більш високому рівні глікемії ШКФ починає лінійно знижуватися. Компенсація вуглеводних порушень призводить до нормалізації ШКФ. У 25% хворих на ЦД 1 типу навіть ідеальна компенсація ЦД супроводжується незначним зниженням ШКФ. Ця когорта хворих належить до групи високого ризику щодо майбутнього розвитку клінічної стадії ДН. Серед причин розвитку стійкої гіперфільтрації при ЦД розглядають різні гормонально-метаболічні фактори: гіперглікемію, гіперкетонемію, збільшення гормона росту і глюкагону, порушення балансу простагландинів, гіперпродукцію передсердного натрійуретичного пептиду, гіперпродукцію ендотеліального фактора релаксації (NО), порушення тубуло-гломерулярного зворотного зв’язку, високе споживання білка. Всі перелічені фактори сприяють підвищенню ШКФ, розширенню приносної артеріоли клубочка нирки, що призводить до збільшення плазмотоку в капілярах клубочка і ШКФ. При цій стадії відзначено і гіпертрофію нирок – збільшення ниркового кровотоку. Медіатори, що спричиняють гіперперфузію нирок і гіперфільтрацію, однакові. На цій стадії ДН фіксуються і структурні зміни нирок тканини: гіпертрофія клубочків і канальців. Гіпертрофія нирок – збільшення нирок у розмірі – спостерігається у 40% хворих на ЦД.
II стадія початкових структурних змін – безсимптомна (латентна) стадія ДН. Екскреція білка із сечею і рівень АТ, як і раніше, залишаються в межах нормальних значень, ШКФ і нирковий кровообіг – підвищені, як і на І стадії ДН. У 5% хворих із ЦД спостерігається ретинопатія. Морфологічна характеристика структурних змін при цій стадії така: спостерігається потовщення базальної мембрани капілярів клубочків і збільшення обсягу мезангінального матриксу.
III стадія розпочинається ДН. Як і попередня стадія, третя характеризується високою ШКФ і ниркового кровотоку, відсутністю білка в загальноклінічному аналізі сечі, нормальним або трохи підвищеним рівнем АТ. Відмітна особливість цієї стадії полягає в появі так званої мікроальбумінурії (МАУ). МАУ називають стан, що характеризується підвищеною екскрецією із сечею альбуміну (в межах 30-300 мг/добу або 20-200 мкг/хв). До клініко-лабораторних особливостей цієї стадії належать підвищення АТ на 10-15%, ретинопатія – у 20-50% хворих, периферична полінейропатія – у 30-50% пацієнтів, загальний аналіз сечі в нормі.
Структурні зміни нирок схожі з попередньою стадією, збільшення мезангінального матриксу >20% від обсягу клубочка. Для виявлення істинної МАУ необхідна наявність багаторазових змін цього показника впродовж декількох тижнів. Якщо у хворих на ЦД виявляють МАУ, то за відсутності належного лікування існує висока ймовірність, що надалі показник екскреції альбуміну із сечею постійно підвищуватиметься і призведе через 8-10 років до протеїнурії. На сьогодні не потребує доказів той факт, що МАУ є не тільки передвісником розвитку клінічної стадії ДН, а й пов’язана з високим ризиком розвитку серцево-судинних ускладнень у цих
хворих. Стійка компенсація вуглеводних порушень, а також застосування препаратів, що нормалізують ниркову гемодинаміку й біохімічний склад базальних мембран клубочків нирок, здатні зупинити патологічний процес у нирках на стадії альбумінурії. Тож дуже важливо своєчасно діагностувати ранню доклінічну стадію ДН і вжити всіх можливих профілактичних заходів для запобігання її подальшому прогресуванню.
IV стадія вираженої ДН характеризується постійною протеїнурією. При цьому екскреція альбуміну із сечею перевищує 200 мкг/хв, або 300 мг/добу, а екскреція білка перевищує 0,5 г (500 мг)/добу. Ця стадія зазвичай проявляється через 15-20 років від початку ЦД. З моменту появи протеїнурії у хворих починається зниження ШКФ зі швидкістю в середньому 1 мл/хв на місяць, що призводить до розвитку термінальної ниркової недостатності. Через 5-7 років від початку протеїнурії розвивається термінальна стадія. Збільшення протеїнурії до 3,5 г/добу і більше може призвести до розвитку класичного нефротичного синдрому, який включає протеїнурію, гіпоальбумінемію, гіперхолестеринемію, появу гіпоальбумінемічних набряків.
Нефротичний синдром при ЦД має низку особливостей:
- набряклість тканин розвивається при більш високому рівні альбумінемії, ніж у хворих із первиннонирковими захворюваннями;
- висока протеїнурія зберігається навіть на стадії ХНН, коли 75% клубочків не працює, тоді як у хворих із первинним гломерулонефритом протеїнурія знижується при розвитку ХНН;
- набряки при ЦД резистентні до терапії діуретиками. Протеїнурія при ДН, як правило, ізольована, тобто не супроводжується зміною осаду сечі (мікрогематурією, лейкоцитурією). Поява еритроцитів і лейкоцитів у сечі хворого на ЦД найчастіше свідчить про приєднання сечової інфекції. Протеїнурична стадія ДН характеризується швидким зростанням рівня АТ. З появою протеїнурії рівень АТ підвищується у 80-90% хворих, а патологічний процес у нирках втрачає свою безпосередню залежність від рівня гіперглікемії, оскільки відсутня кореляція між темпом зниження ШКФ і рівнем глікозування гемоглобіну [13].
Припускають, що факторами, які прискорюють розвиток ХНН у хворих на ЦД, є: початок діабету в пубертатному віці (50%); наявність АГ; гіперліпідемія (гіпертригліцеридемія); наявність набрякового синдрому; низький гематокрит; спадкова обтяженість за ЦД; незадовільна компенсація ЦД. Сукупність перелічених клінічних і лабораторних показників призводить до швидкого розвитку ХНН при ДН і вказує на необхідність більш агресивного лікування діабетичного ураження нирок. Протеїнурична стадія ДН характеризується наростанням тяжкості та інших мікро- і макросудинних ускладнень ЦД: ретинопатії – у 100% хворих, полінейропатії – у 100%, автономної нейропатії (ортостатизм, ентеропатія), гіпертензії – у 80-90% хворих, гіперліпідемії – у 60-80%, ішемічної хвороби серця – у 50-70% хворих. При цьому у 70% випадків виявляються найбільш тяжкі стадії ДР – препроліферативна і проліферативна, що призводять до втрати зору. Таке співдружне прогресування ДР і нефропатії дало змогу клініцистам виділити симптомокомплекс, що дістав назву нирково-ретинальний синдром. Зміни на очному дні завжди спостерігаються раніше, ніж з’являється протеїнурія. На стадії протеїнурії досить швидко прогресує і ДН – як периферична, так і автономна її форми. Це ще більше погіршує стан хворих із ДН. Протеїнурична стадія ДН супроводжується розвитком серцево-судинної патології. Цей взаємозв’язок можна зрозуміти: втрата білка із сечею призводить до надмірного синтезу атерогенних ліпідів. Регрес функціональних і структурних змін нирок на цій стадії ДН неможливий. Протеїнурична стадія ДН незворотня. Можна тільки сповільнити прогресування ДН до стадії ниркової недостатності, застосувавши тактику інтенсивної антигіпертензивної терапії.
V стадія (уремічна) – стадія хронічної ниркової недостатності.
На цій стадії фільтраційна функція нирок продовжує знижуватися, що призводить до наростання концентрації у крові токсичних азотистих шлаків. Біохімічними критеріями зниженого ШКФ є підвищення креатиніну, сечовини сироватки крові, гіперкаліємія і гіперфосфатемія. На цій стадії спостерігаються структурні зміни (склероз >80% клубочків, артеріологіаліноз, тубулоінтестиціальний фіброз), що ведуть до зниження ШКФ. Морфологічно це визначається як тотальний дифузний або вузликовий гломерулосклероз. На стадії ХНН може фіксуватися зниження протеїнурії, що вважається несприятливою прогностичною ознакою, яка свідчить про прогресуючу оклюзію ниркових клубочків. Термінальна стадія ураження нирок характеризується значним зростанням АТ, іноді тяжко керованого; приєднується затримка рідини з розвитком набрякового синдрому, резистентного до сечогінних препаратів; розвиваються симптоми серцевої недостатності, яка може ускладнитися розвитком набряку легень. На стадії ХНН різко прогресують й інші мікро- і макросудинні ускладнення ЦД: усі 100% хворих із ХНН мають ДР, із них 80% – препроліферативну і проліферативну стадії, що призводить до втрати зору в 30-40% хворих; усі хворі з ХНН мають периферичну полінейропатію, яка посилюється при уремії внаслідок специфічного ураження нервових волокон; посилюється автономна нейропатія, виявляється ортостатична гіпотензія, що ускладнює підбір адекватної дози антигіпертензивних препаратів; інфаркти міокарда та інсульти є основною причиною смерті хворих на ЦД на стадії ХНН, які отримують лікування гемодіалізом. На стадії уремії приєднуються і специфічні симптоми, характерні для хворих із вираженою втратою фільтраційної функції нирок: ниркова анемія (нормохромна і нормоцитарна), яка розвивається унаслідок порушення синтезу еритропоетину в нирках; ниркова остеодистрофія – ураження скелета і м’яких тканин, що розвиваються внаслідок порушення фосфорно-кальцієвого обміну. Для ХНН характерні гіперфосфатемія і гіпокальціємія, які призводять до гіперплазії паращитовидних залоз і розвитку вторинного гіперпаратиреозу. Гіперпаратиреоз виникає при помірному зниженні ШКФ до 60-40 мл/хв, а у разі зниження фільтраційної функції нирок до 40-20 мл/хв розвиваються ознаки остеомаляції. При зниженні ШКФ<15 мл/хв розвивається уремічна інтоксикація організму азотистими шлаками (свербіж, нудота, блювота) та явища метаболічного ацидозу, що викликають дихання Кусмауля. В таблиці 2 наведені обов’язкові та додаткові методи досліджень, що проводяться пацієнтам із ДХН.

Альбумінурія – маркер нирково-гломерулярних захворювань, високого серцево-судинного ризику і перших клінічних проявів діабетичної хвороби нирки (ДХН) [14]. Не в усіх хворих із ДХН і зниженою ШКФ спостерігається альбумінурія. З іншого боку, зниження ШКФ<60 мл/хв/1,73 м2 у хворих на ЦД без альбумінурії може бути проявом ХХН, яка виникла через іншу етіологічну причину, а не внаслідок розвитку ДХН, тож потребує уточнення причини ХХН.
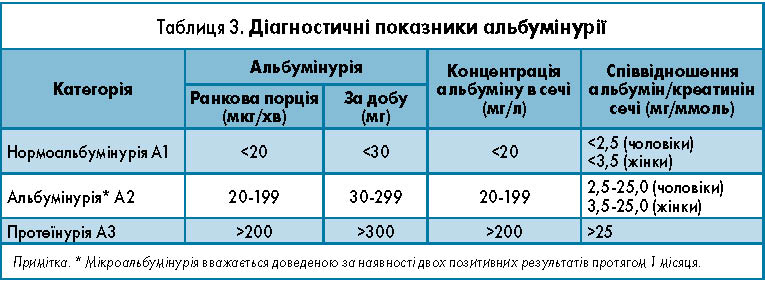
Слід ураховувати й велику варіабельність результатів альбумінурії (близько 40%). Для зменшення випадковості визначення альбумінурії в разовій або добовій сечі загальноприйнятим діагностичним тестом стало вимірювання альбумін-креатинінового співвідношення, яке дозволяє використовувати однократну ранкову порцію сечі [15]. Діагностичні показники альбумінурії наведені в таблиці 3.
Розвиток ДН у хворих на ЦД не тільки погіршує прогноз, а й потребує корекції застосованої цукрознижувальної терапії, специфічної профілактики ДН та лікування власне проявів-ускладнень ДН. Своєчасне виявлення ДН можливе лише при скринінговій оцінці функції нирок у хворих на ЦД за ШКФ. ДХН встановлюють на підставі зниження ШКФ<60 мл/хв/1,73 м2 і ураження нирки за рівнем екскреції альбуміну, оціненого за співвідношенням альбумін/креатинін у сечі >30 мг/г [15].
У клінічній практиці використовують не вимірювання, а обчислення ШКФ. Для цього застосовують формули кліренса ендогенних фільтраційних маркерів (кретинін і цистатин С) [16]. На визначення ШКФ за рівнем ендогенних фільтраційних маркерів впливають безліч факторів, тому формули обчислення дають велику погрішність, особливо при високій ШКФ [16]. Для обчислення ШКФ рекомендовано використовувати розроблені формули.
Методи обчислення ШКФ
- Формула Кокрофта-Голта *:
ШКФ (мл/хв) = [(140-вік (роки)) × маса тіла (кг)] / 72 × креатинін сироватки (мкмоль/л) × 0,85 (для жінок). - Формула MDRD:
ШКФ (мл/хв/1,73 м2) = 186 × (креатинін сироватки) –1,154 × (вік)) –0,203 × 0,742 (для жінок) × 1,210 (для представників негроїдної раси). - Формула CKD-EPI:
СКФ (мл/хв/1,73 м2) = 141 × min (креатинін сироватки / k, 1) а × max (креатинін
сироватки / k, 1) –1,209 × 0,993 вік × [1,08 для жінок] × 1,159 [для представників негроїдної раси],
де k – 0,7 для жінок і 0,9 для чоловіків,
а – (–0,329) для жінок і (– 0,411) для чоловіків. - Формула Швартца (для дітей)*:
СКФ (мл/хв) = 4,3 × зростання (м) / креатинін сироватки. - Формула Коунахана (для дітей):
СКФ (мл/хв/1,73 м2) = 3,8 × зростання (м) / креатинін сироватки.
Величину СКФ, обчислену за формулами Кокрофта-Голта та Швартца, необхідно приводити до стандартної площі поверхні тіла 1,73 м2.
У 2004 році цистатин С був офіційно визнаний Управлінням з контролю за продуктами харчування та лікарськими препаратами США (FDA) як маркер для альтернативного визначення ШКФ. Рекомендовано такі формули для обчислення ШКФ за одноразовим визначенням цистатину С у сироватці крові.
- ШКФ = 91,627* цистатин С; ШКФ = 80,35 / цистатин С – 4,32.
- ШКФ = 78 / цистатин С + 4; ШКФ = 119 / цистатин С – 33.
- ШКФ = 100 / цистатин С – 14.
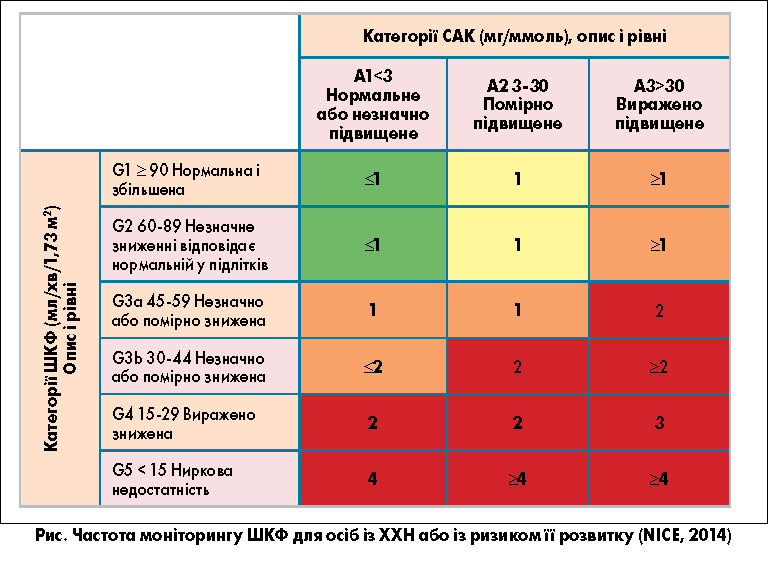
Визначення сироваткового рівня цього показника може бути корисним для скринінгу ранніх стадій ХХН у хворих на ЦД [17]. Частота моніторингу ШКФ для осіб із ХХН або із ризиком її розвитку подано на рисунку.
На підставі рівня ШКФ визначають стадію ХХН, етіологічним чинником якої є ДН, що разом свідчить про ДХН.
Рекомендовано класифікацію ХХН на основі значень ШКФ і альбумінурії [16].
Первинне обстеження на наявність ДХН слід проводити відразу після встановлення діагнозу ЦД 2 типу і через 5 років від початку ЦД 1 типу [15]. За відсутності у хворого ДХН скринінгове визначення альбумінурії і ШКФ слід проводити щорічно (табл. 4).
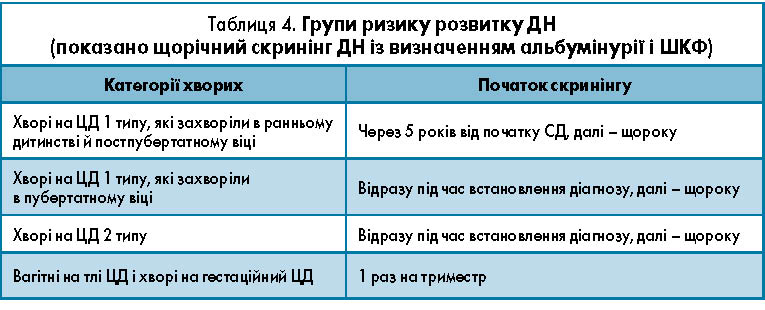
Діагноз ДН відповідно до класифікації ХХН
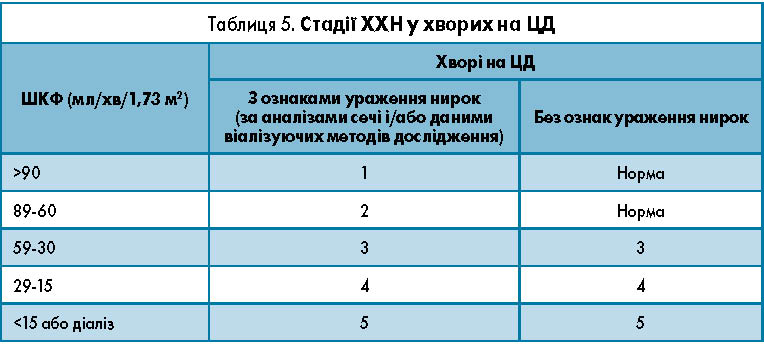
У разі виявлення у хворого на ЦД альбумінурії або протеїнурії встановлюють діагноз з уточненням стадії ХХН (залежно від ШКФ, табл. 5): ДН, стадія МАУ, ХХН 1, 2, 3 або 4; ДН, стадія протеїнурії, ХХН 1, 2, 3 або 4; ДН, ХХН 5 (лікування замісної ниркової терапії). У разі виявлення у хворого ЦД зниження ШКФ<60 мл/хв і за відсутності інших ознака ураження нирок (альбумінурії, протеїнурії) встановлюють діагноз: ХХН 3 або 4; ХХН 5 (лікування замісною нирковою терапією).
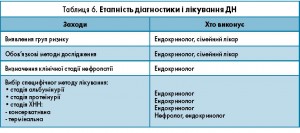
Необхідність виявлення ДХН у хворих ЦД зумовлено не тільки зміною прогнозу, а й необхідністю корекції терапії та лікування ускладнень, таких як анемія, порушення фосфорно-кальцієвого обміну й підготовка до сучасної замісної терапії. Етапність діагностики і лікування ДН подано у таблиці 6.
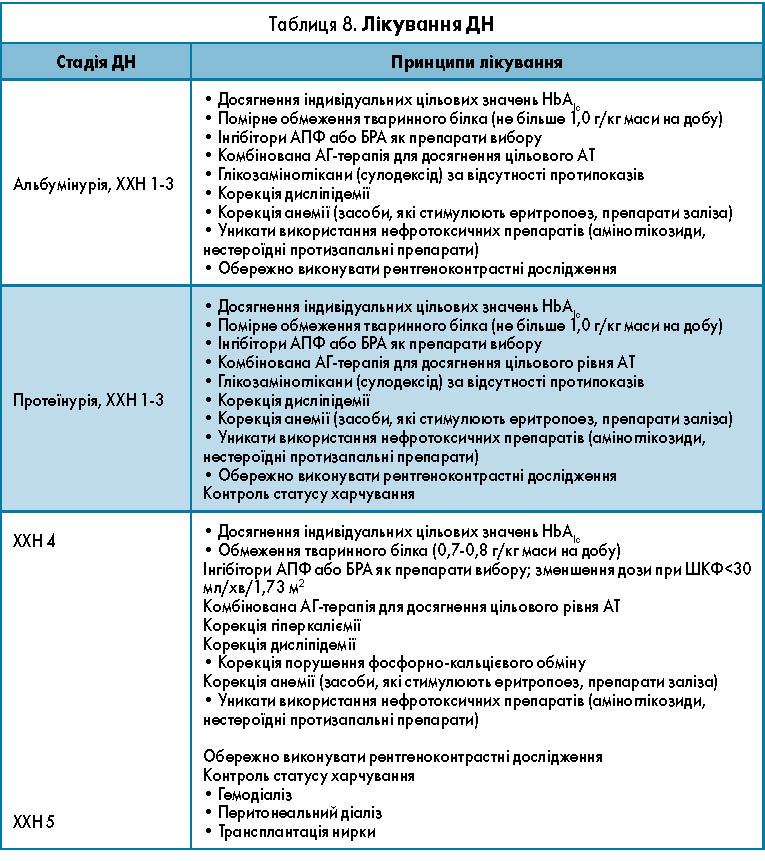
Комплексна терапія діабетичного ураження нирок передбачає кілька напрямів.
І. Компенсація вуглеводного обміну. Одним із критеріїв оптимальної компенсації є рівень НbА1с<7,5%, що дає змогу запобігти розвитку ДН і значною мірою знизити темп її прогресування. Однак при зниженні ШКФ існування еритроциту вкорочене, що призводить до зниження НbА1с [18]. При розвитку анемії у хворих із ДХН коректність оцінки визначення рівня НbА1с додатково знижується, тож визначення рівня глікемії для вирішення питання корекції терапії є приорітетним. У хворих із ДХН значно зростає ризик гіпоглікемії. Загалом у хворих із ЦД і ДХН рекомендоване зниження рівня НbА1с до 7% для запобігання прогресуванню мікросудинних ускладнень ЦД, проте не нижче 7%. У пацієнтів із супутньою серцево-судинною патологією і ризиком гіпоглікемій цільовий рівень НbА1с має становити >7%. При ШКФ<60 мл/хв/1,73 м2 у разі ризику гіпоглікемії рекомендовано контроль глікемії з НbА1с<8% [15].
Препаратом вибору при лікуванні ЦД 2 типу є метформін. Метформін застосовують без корекції дози до ШКФ 45 мл/хв/1,73 м2, у разі зниження ШКФ<45 мл/хв/1,73 м2 необхідно зменшити дозу в 2 рази – до 1000 мг/добу [15]. При зниженні ШКФ<30 мл/хв/1,73 м2, а також при гострому ураженні нирок, інфаркті міокарда метформін слід відмінити. Цукрознижувальні препарати, які застосовуються у хворих із ХНН, подано у таблиці 7.

Адекватну інсулінотерапію призначає ендокринолог. Найбільшого поширення на сьогодні отримала інтенсифікована (або базис-болюсна) схема людськими генно-інженерними інсулінами та інсуліновими аналогами.
Лікування інсуліновою помпою (НПІІ – безперервна підшкірна інфузія інсуліну, CSII – continuous subcutaneous insulin infusion) – набагато дорожчий метод, аніж традиційне лікування шприцами або шприц-ручками. Використання помпи не відміняє інтенсивну інсулінотерапію, а допомагає вдосконалювати її, щоб доводити контроль вуглеводного обміну до цільових рівнів. Слід зазначити, що важливе значення в підтримці задовільної компенсації ЦД мають самоконтроль і навчання в Школі діабету.
II. Корекція внутрішньоклубочкової гіпертензії – найважливіший компонент патогенетичного лікування ДН. Для цього призначають інгібітори ангіотензинперетворювального ферменту (ІАПФ). Згідно з літературними джерелами своєчасне і правильне призначення ІАПФ дає змогу значно знизити швидкість прогресування ДН. Слід зазначити, що ІАПФ у стадії протеїнурії рекомендований для постійного застосування. Препарати цієї групи широко використовують при ДН. В останні роки в літературі дедалі частіше згадують інші групи антигіпертензивних препаратів, у тому числі антагоністи рецепторів ангіотензину II 1 типу, як засоби корекції внутрішньоклубочкової і системної гіпертензії при ДН.
III. У клінічних дослідженнях продемонстрована доцільність застосування в лікуванні початкової ДН препаратів глікозаміногліканів, які мають здатність запобігати проліферації мезангія і гіперпродукції екстрацелюлярного матриксу, а також потовщення базальної мембрани клубочків і порушення її проникності й зарядоселективності.
IV. На сьогодні дискутується питання про обмеження тваринного білка в лікуванні ДН. Ці обмеження мають на меті зниження гемодинамічного навантаження на нирки, що провокується високобілковою дієтою, і зменшення фільтраційного навантаження білком на нирки. Окремі автори вважають недоцільним, особливо у дітей і підлітків, обмежувати білок як основний будівельний матеріал організму, що росте. В рекомендаціях ISPAD Consensus (2000) запропоновано використовувати дієту з помірним обмеженням білка (до 0,9-1,2 г/кг/добу) підліткам на стадії МАУ, а на стадії протеїнурії необхідно обмежувати білок до 0,8-0,9 г/кг/добу.
V. Гіполіпідемічна терапія (секвестранти жовчних кислот, нікотинова кислота та її похідні, фібрати, інгібітори ГМК-КоА-редуктази) – важливий компонент патогенетичного лікування у дорослих пацієнтів із ДН у стадії вираженої нефропатії, що поєднується із дисліпідемією. У дітей і підлітків із ДН у разі виявлення ліпідних порушень допустимо використовувати секвестранти жовчних кислот (холестирамін, холестинол).
VI. Наразі одним із перспективним напрямів патогенетичної терапії ДН розглядається питання корекції підвищеного рівня гомоцистеїну із застосуванням препаратів фолієвої кислоти й вітамінів групи В (у дітей та підлітків).
VII. Лікування супутньої патології сечової системи – окрема складова терапії, спрямованої на запобігання розвитку гломерулосклерозу при ЦД.
Принципи лікування ДН залежно від стадії вказані в таблиці 8.
VIII. Показами до початку замісної ниркової терапії у хворих на ЦД і ХНН є:
- ШКФ <15 мл/хв/1,73 м2.
- Гіперкаліємія (>6,5 мекв/л), що не коригується консервативними методами лікування.
- Тяжка гіпергідратація з ризиком розвитку набряку легенів.
- Наростання білково-енергетичної недостатності.
Цільові значення НЬА1с у хворих на ЦД на діалізі:
- у хворих без виражених серцево-судинних ускладнень <7,5%;
- у хворих із тяжкими судинними ускладненнями та схильністю до гіпоглікемії: 7,5-8,0%.
Контроль рівня АТ у хворих на ЦД на діалізі:
- Цільовий рівень АТ у хворих ЦД до діалізу і між сеансами діалізу відрізняється від рекомендованого для всієї популяції діалізних хворих загалом (130/80-140/90 мм рт. ст.).
- Препаратами першого ряду (як і в додіалізному періоді) залишаються ІАПФ і БРА. Ниркова анемія у хворих на ЦД на діалізі
- Цільовий рівень гемоглобіну крові >11,0 г/дл, проте <12,0 г/дл.
- Лікування засобами, що стимулюють еритропоез (еповітан, епоетин-альфа), і препаратами заліза (пероральними й парентеральними).
На завершення слід зазначити, що своєчасна діагностика та лікування ДН багато в чому визначають якість життя і прогноз пацієнта із ЦД. Стримування темпів прогресування діабетичного ураження нирок найбільш перспективно на ранніх стадіях. Надалі своєчасне проведення лікувально-діагностичних заходів допоможе забезпечити зменшення кількості пацієнтів, які потребують замісної діалізної терапії, знизити ризик серцево-судинної патології, асоційованої із ХНН.
Література
- Bernard С. Compte Rendu de la Sosiete du Biologie. 1849; 1: 80-81.
- Ross J., Goldmann G.K. Effect of streptozotocin-induced diabetes of kidney weight and compensatory hypertrophy in the rat. Endocrinology. 1971; 88; 1079-1082.
- Mogensen C.E., Andersen M.J. Increased kidney size and glomerular filtration rate in untreated juvenile diabetics. Normalisation by insulin treatment. Diabetologia, 1975; 11: 221-224.
- Шестакова M.B. Диабетическая нефропатия: фатальное или предотвратимое осложнение? Рус. мед. журнал, http:// www.rmj.ru/ 2007/3.
- Gilbert R., Tsalamandris С., Allen T. et al. Early nephropathy predicts vision threatening retinal disease in patients with type 1 diabetes mellitus. J. Am. Sos. Nephrol. 1998 (9): 85-89.
- Hamman R.E. Epidemiology of microvasenlar complicatins // In: International Textbook of Diabetes Mellitus. Ed. by R.G. M.M. Alberti., P. Zimmet, R.A. De Fronzo. – John Wiley, Sons Ltd. – 1997. – № 2. – P. – 1293-1313.
- Gall M.A., Rossing P., Skott P., Damsbo P. et al. Prevelence of micro- and macroalbuminuria, arterial hypertension, retinopathy and large vessel disease in European type 2 (non-insulindependent) diabetic patients // Diabetologia. – 1991. – Vol. 34. – P. 655-661.
- Шамхалова М.Т. и др. Терапевтический архив 2006 г. – № 10. – С. 27-33. Поражение почек при сахарном диабете 2 типа.
- Postma M.J., de Zeeww D. The economic benefits of preventing end – stage renal disease in patients with type 2 diabetes mellitus // Nephrol Dial. Tsansplant. – 2009. – № 24. – P. 2475-2980.
- Villar E., Chang S.H., Me Donald S.P., Incidences, Treatments, Outcjmes, Sex Effect on Survival in Patients With End – Stage Renal Disease by Diabetes Status in Australia and New Zealand (1991-2005) // Diabetes Care. – 2007. – № 30 (12). – P. 3070-3076.
- NKF-K/DOKI clinical practice guidelines for chronic kidney diseases: evaluation, classification and stratification // Am. J. Kidney Dis. – 2002. – 39(suppl.1). – S. 17-31.
- Mogensen C.E. The Diabetic Kidney. New Jersey: Нumana Press. 2006.
- Viberti G.V. 1992.
- Bakris G.I., Molitch M. Microalbuniminuria as risk predictor in diabetes: the contimuing saga. Diabetes Care. 2014; 37: 867-875.
- Diabetic Kidney Disease: A Report Form an ADA Consensus Conference.Am J Kidney Dis. 2014; 64(4): 510-553.
- Evaluation and management of chronic kidney disease: synopsis of the kidney disease improving global outcomes 2012 clinical practice guideline. Ann Inter.Med. 2013 Jun 4; 158 (11): 825-30.
- Urinary cystatin C as a specific marker of tubular disfunction [Text] / M. Conti, S. Moutereau, M. Zater [et al.] // Clin. Chem. Lab. Med. – 2006. – Vol 44 (3). – P. 288-291.
- Vos F.E., Schollum J.B., Coulter C.V., Doyle T.C.A. Dufful S.B., Walker R.J. Red blood cell survival in long-tern dialysis patiets. Am.J.Kidney Dis. 2011; 58: 591-598.
- Ellis D., Lloyd C., Becker D.J. et al. The changing course of diabetic nephropathy: Low-density lipoprotein cholesterol and blood pressure correlate with regression of proteinuria // Am. J. Vid. Dis. – 1996. – Vol. 27. – P. 809-818.