


7 листопада, 2017
Комплексна патофізіологія набутої апластичної анемії
Апластична анемія (АА) – синдром недостатності кісткового мозку, що характеризується аплазією кісткового мозку і панцитопенією в периферичній крові. У пацієнтів з АА часто спостерігаються симптоми анемії, пурпура і геморагії, а також інфекції. Найчастіше АА є набутою й ідіопатичною. Хоча патофізіологія АА є не повністю зрозумілою, той факт, що 80% пацієнтів відповідають на імуносупресивну терапію (ІСТ) антитимоцитарним глобуліном і циклоспорином, вказує на роль імунних механізмів у розвитку захворювання. У багатьох пацієнтів ІСТ дозволяє досягти тривалої виживаності без подій, проте ризик рецидиву, клональної еволюції в мієлодиспластичний синдром або мієлолейкоз залишається високим. У випадках тяжкої АА в дітей і дорослих молодого віку радикальним методом лікування є трансплантація гемопоетичних стовбурових клітин (ГСК). Наявні лабораторні та клінічні дані свідчать про те, що в патогенезі набутої АА (НАА) важливу роль відіграє імуноопосередковане пригнічення гемопоезу. Проте в нещодавніх дослідженнях було встановлено, що гемопоетичні стовбурові клітини / клітини-попередники (ГСК/КП) і мезенхімальні стовбурові клітини кісткового мозку (МСК-КМ), отримані від пацієнтів з НАА, мають первинний дефект, який робить ці клітини вразливими в умовах недостатності кісткового мозку. У цьому огляді обговорюються останні досягнення в патофізіології НАА з акцентом на дизрегульовані імунні відповіді.
Дизрегульовані відповіді Т-клітин
Експериментальні і клінічні дані свідчать про важливу роль CD8+ Т-клітин (цитотоксичних Т-лімфоцитів, Т-кілерів) у патогенезі НАА. У ранніх дослідженнях було встановлено, що лімфоцити кісткового мозку і периферичної крові пацієнтів з НАА здатні пригнічувати гемопоез in vitro. У подальшому виявилося, що лімфоцитами, що пригнічують гемопоез, є CD8+ олігоклональні (тобто з високообмеженою різноманітністю рецепторів Т-лімфоцитів – TCR) T-клітини. У пацієнтів, які відповіли на ІСТ, знижуються кількість імунодомінантних клонів і варіабельність TCR. У момент рецидиву захворювання початкові патогенні домінантні клони з’являються знову, іноді разом з новими клонами, що корелює з розширенням епітопу імунної відповіді. У деяких випадках клони під час ремісії персистують, що свідчить про толерантність Т-клітин.
Причини активації Т-клітин при НАА не встановлені. Підвищена презентація певних алелів HLA вказує на роль розпізнавання антигенів. Аберантна експресія генів, які відповідають за сигнали TCR, може сприяти дисфункції Т-клітин при НАА. Зокрема, у пацієнтів з НАА значно підвищується експресія генів CD3γ, CD3δ, CD3ε і CD3ζ порівняно зі здоровими пацієнтами. Підвищена експресія цих генів при НАА свідчить, що периферичні Т-клітини можуть піддаватися постійній стимуляції, і це призводить до патологічної активації Т-клітин.
У пацієнтів з НАА підвищується кількість і/або функція CD4+ Т-клітин (Т-хелперів), зокрема таких, що продукують інтерферон-γ – IFNγ (Th1-клітини), інтерлейкін – IL-4 (Th2-клітини), IL-17 (Th17-клітини), а також Treg. Giannakoulas і співавт. (2004) встановили, що пацієнти з нелікованою або рефрактерною НАА мають значно підвищену частку нестимульованих Тh1-клітин, які продукують IFNa і IL-2, за нормальної кількості Th2-клітин; це призводить до зсуву співвідношення IFNγ/IL-4 у бік Th1-відповіді. У пацієнтів у ремісії також спостерігається підвищення частки Th1-клітин з паралельним збільшенням кількості Th2-клітин і нормальним співвідношенням IFNγ/IL-4.
Клональність Th1-клітин, як і CD8+ T-клітин, є обмеженою, що свідчить про антигенобумовлену експансію Th1. Функціонально ці домінуючі CD4+ Th1-клони при НАА секретують IFNγ і фактор некрозу пухлини (TNF), здатні викликати лізис аутологічних CD34+ клітин й інгібувати формування їх гематопоетичних колоній.
Регуляторні Т-лімфоцити – це CD4+ CD25+ FoxP3+ клітини, що відіграють фундаментальну роль в аутоімунітеті. Людські CD4+ CD25+ FoxP3+ клітини складаються з трьох фенотипово й функціонально відмінних популяцій: CD45RA+FoxP3low Treg-клітин у стані спокою (rTreg) й активованих CD45RA-FoxP3high (aTreg), які in vitro проявляють супресивну дію, і цитокінсекретуючих CD45RA-FoxP3low несупресивних Treg-клітин. Співвідношення і функції цих субпопуляцій Treg варіюють при різних аутоімунних хворобах. У пацієнтів з НАА кількість Treg корелює з тяжкістю захворювання; крім того, спостерігається відносне зниження rTreg і aTreg з підвищенням цитокінсекретуючих не-Treg-клітин. Функціонально Treg у пацієнтів з НАА мають первинний дефект, а саме порушену здатність до міграції через знижену експресію CXCR4 і зменшену здатність пригнічувати нормальні функції ефекторних Т-лімфоцитів, зокрема продукцію IFNγ. Пацієнти з більшою кількістю Treg краще відповідають на ІСТ.
Th17-клітини – CD4+ Т-лімфоцити, що секретують IL-17A, – цитокін, який координує запалення тканин шляхом індукування цитокінів (CXCL8, CXCL6, CXCL1), факторів росту (гранулоцитарного колонієстимулюючого і гранулоцитарно-макрофагального колонієстимулюючого факторів) і молекул адгезії (молекула міжклітинної адгезії 1), що посилює накопичення нейтрофілів і гранулопоез. IL-17 пов’язаний з багатьма аутоімунними хворобами, зокрема ревматоїдним артритом і запальними захворюваннями кишечнику, проте роль цього цитокіну при НАА вивчена недостатньо. Відомо, що в більшості пацієнтів з НАА плазмові рівні IL-17 не підвищуються. У деяких дослідженнях експансія популяції Th17-клітин корелювала з виснаженням натуральних Treg у крові, що вказує на зворотний зв’язок між Th17 / CD4+CD25highFoxP3+ Treg й аутоімунним процесом при НАА.
Загалом наявні дані свідчать, що поєднання експансії Th1, Th2 і, можливо, Th17 зі зниженими або порушеними імунофенотипом і функцією Treg бере участь у патогенезі НАА, особливо тяжких і дуже тяжких форм. Експансія Th1-клітин, імовірно, є антигенопосередкованою, Th17-клітини можуть безпосередньо або опосередковано модулювати Th1-клітини, а присутність дисфункціональних Treg-клітин може посилювати цю аутоімунну відповідь. Специфічні аутоантигени, які спричиняють аберантні імунні відповіді проти ГСК/КП при НАА, дотепер не встановлені.
Вроджений імунітет
Більшість досліджень НАА були спрямовані на вивчення Т- і В-клітин, проте останнім часом все більше доказів свідчать, що в патогенезі захворювання також може брати участь дисфункціональний вроджений імунітет.
У низці досліджень було встановлено, що в пацієнтів з НАА зменшуються кількість і активність натуральних кілерів (NK), а відновлення цитотоксичності NK-клітин корелює з відновленням гемопоезу після ІСТ. Дефіцит цитолітичної активної NK може бути наслідком мутації гена перфорину або пригнічення аутологічних гранулоцитів.
У пацієнтів з НАА визначається дефіцит стресіндуцибельних лігандів NKG2D, зокрема UL16-зв’язувальних білків ULBP1, ULBP2 і ULP3, а також білка MICA на гранулоцитах і клітинах кісткового мозку, при цьому рівень експресії цих лігандів позитивно корелює з недостатністю кісткового мозку й адекватною відповіддю на ІСТ. Ідентифікація лігандів NKG2D може бути корисною в ранній діагностиці імуноопосередкованого ураження кісткового мозку і прогнозуванні відповіді на ІСТ.
Мієлосупресивні цитокіни
Аутореактивні Т-клітини в пацієнтів з НАА секретують прозапальні цитокіни, зокрема IFNγ і TNF, що призводить до підвищення рівнів цих цитокінів у кістковому мозку і периферичній крові. IFNγ і TNF знижують формування колоній гемопоетичних клітин-попередників внаслідок індукування апоптозу CD34+ клітин Fas-Fas-лігандним та/або TNF-обумовленим апоптозіндукуючим лігандним (TRAIL) шляхом.
Присутність внутрішньоклітинного IFNγ у Т-клітинах у крові і кістковому мозку пацієнтів з НАА може слугувати предиктором відповіді на ІСТ, а також індикатором розвитку рецидиву. Натомість внутрішньоклітинна експресія TNF у Т-клітинах кісткового мозку асоціюється з негативними клінічними результатами.
Трансформуючий фактор росту β1 (TGFβ1) – ще один багатофункціональний цитокін, який бере участь у гемопоезі. У пацієнтів з НАА часто визначаються генотипи, які асоціюються з високою продукцією TGFβ1, особливо генотип -509 ТТ, проте в деяких випадках може спостерігатися зниження рівнів TGFβ1 у крові і кістковому мозку. Такі поліморфізми TGFβ1, як -590 C/T rs1800469 і P10L C/T rs1800470, не впливають на схильність до НАА, хоча можуть асоціюватися з кращою відповіддю на ІСТ.
Гемопоетичні стовбурові клітини / клітини-попередники
Нещодавні дослідження свідчать, що в певних підгрупах пацієнтів з НАА до основного патофізіологічного механізму захворювання – імуноопосередкованого руйнування ГСК/КП – може приєднуватись ще один механізм, а саме первинний дефект ГСК/КП.
Так, у деяких пацієнтів з НАА, особливо в тих, які не відповідають на ІСТ, спостерігається значне зменшення довжини теломер в лейкоцитах. Мутації в генах теломеразного комплексу, зокрема теломеразного РНК-компонента (TERC) і теломеразної зворотної транскриптази (TERT), порушують здатність підтримувати довжину теломери, що призводить до зниження здатності до проліферації і виживаності гемопоетичних клітин та, як наслідок, до зменшення пулу ГСК. У разі дії зовнішнього чинника, що викликає імуноопосередковане руйнування ГСК, пацієнти з такими мутаціями можуть бути більш вразливими до розвитку недостатності кісткового мозку і неадекватно відповідати на ІСТ.
Проте більшість хворих на АА зі скороченими теломерами не мають відомих мутацій, які відповідають за таке скорочення, що вказує на залучення інших генів та/або зовнішніх факторів. Прискорене скорочення теломер у деяких нелікованих або рефрактерних до лікування пацієнтів з НАА може бути наслідком підвищеної проліферації ГСК/КП, подібно до такої в пацієнтів після алогенної ТГСК, коли обмежений клональний гемопоез і «регенеративний стрес» призводять до хромосомної нестабільності ГСК/КП. При цьому менша довжина теломер корелює зі стійкою цитопенією після ІСТ, підвищеним ризиком рецидиву, клональною еволюцією, моносомією 7 і зниженою загальною виживаністю.
Незалежно від етіології зменшення довжини теломер, у пацієнтів з НАА більш короткі й дисфункціональні теломери не лише обмежують проліферацію нормальних ГСК/КП, а й спричиняють хромосомну нестабільність і схильність до злоякісної трансформації. При НАА критично мала довжина теломер може слугувати біомаркером клональної еволюції до моносомії 7.
У пацієнтів з НАА CD34+ ГСК/КП демонструють значно знижену регуляцію генів контрольних точок клітинного циклу, зокрема генів, які кодують циклінзалежну кіназу 6 (CDK6), CDK2, цикліни Е і А, комплементарну групу G анемії Фанконі (FANCG), генів c-myb і c-myc.
Це може пояснювати нездатність ГСК/КП до повної реплікації і компенсації імуноопосередкованого пошкодження, а також появу передракових або анеуплоїдних клітин у пацієнтів з АА, які залишаються вразливими до розвитку мієлодисплазії та мієлолейкозу навіть через багато років після гематологічного відновлення завдяки ІСТ.
У багатьох пацієнтів з НАА визначаються набуті соматичні мутації в генах, пов’язаних з мієлоїдними злоякісними процесами, зокрема ASXL1, DNMT3A, TET2 і BCOR; наявність таких мутацій є предиктором високого ризику трансформації у мієлодиспластичний синдром або гострий мієлолейкоз.
Мезенхімальні стовбурові клітини кісткового мозку
МСК-КМ є ключовими клітинами-попередниками мікросередовища кісткового мозку, які відіграють важливу роль у довгостроковому підтриманні гемопоезу. МСК-КМ диференціюються в низку стромальних клітин, що становлять нішу ГСК. Свої функції МСК-КМ і диференційовані стромальні пухлини здійснюють шляхом продукції і секреції цитокінів, хемокінів і молекул екстрацелюлярного матриксу. МСК чинять виражені імуносупресивні ефекти на Т-клітини, NK і антигенпрезентуючі клітини, тож вони можуть залучатися до патогенезу АА.
У низці досліджень було продемонстровано, що МСК з апластичного кісткового мозку мають первинні та/або вторинні дефекти, зокрема знижений проліферативний і клоногенний потенціал, підвищений апоптоз, аберантну диференціацію, а також неадекватне пригнічення активації Т-клітин, продукції TNF і IFNγ. Цікаво, що функціональний дефіцит МСК-КМ щодо зниження праймування, проліферації і вивільнення цитокінів Т-клітин персистує нескінченно довго після ІСТ, проте може нормалізуватися після трансплантації кісткового мозку.
Профілювання експресії генів МСК-КМ, отриманих від пацієнтів з НАА, дозволило визначити значну різницю в експресії великої кількості генів, залучених до клітинного циклу, поділу клітин, хемотаксису, передачу сигналів адипогенезу й диференціації ГСК. Наприклад, у пацієнтів з АА експресія гена, що кодує GATA-зв’язувальний протеїн 2 (GATA2; транскрипційний фактор, необхідний для утворення і нормального функціонування ГСК/КП), знижується не лише в ГСК/КП, а й у МСК-КМ.
У дітей з АА також спостерігається значне зниження експресії CXCL12 у МСК-КМ, що асоціюється зі зниженням виживаності і потенціалу до диференціації цих клітин. Одночасна трансплантація ГСК/КП й алогенних МСК, отриманих з кісткового мозку або пуповинної крові, зменшувала ризик хвороби «трансплантат проти хазяїна» і покращувала стромальну функцію в пацієнтів з АА. Ці дані не лише підтверджують імуномодулюючі властивості МСК, а й надають непрямі докази того, що МСК-КМ можуть брати участь у патогенезі АА.
Висновки
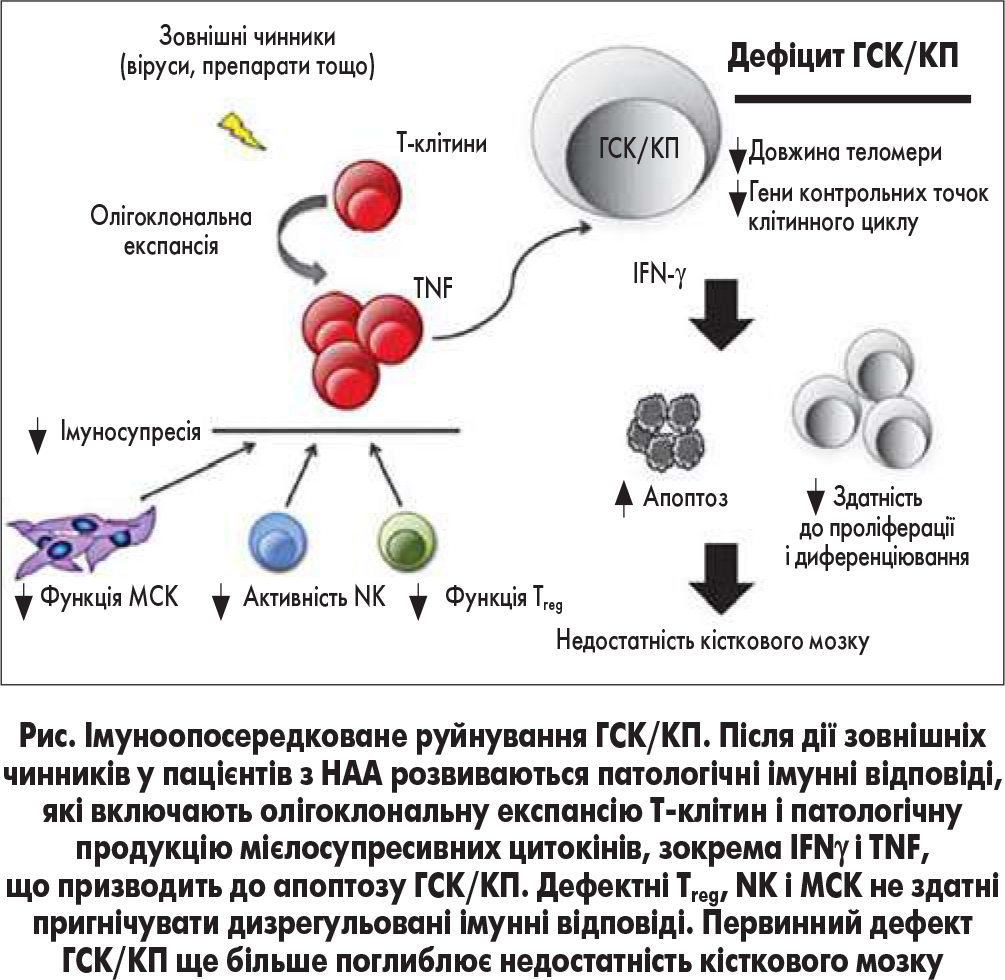
Патологічно змінена імунна регуляція при НАА, що залучає CD8+ Th1, Treg і клітини вродженого імунітету, призводить до деструкції ГСК/КП (рис.). Антигени, які беруть участь у цьому процесі, дотепер не встановлені. Первинний дефект ГСК/КП, присутній у пацієнтів з НАА, поглиблює недостатність кісткового мозку. Хоча сучасне лікування дозволяє значно поліпшити прогноз захворювання, для кращого розуміння патофізіологічних механізмів і створення більш ефективних препаратів необхідні подальші дослідження.
Список літератури знаходиться в редакції.
Zeng Y., Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol, 2015 Jun; 180 (3): 361-70.
Переклав з англ. Олексій Терещенко
Тематичний номер «Онкологія» № 4 (50), жовтень 2017 р.