


21 жовтня, 2019
Електрокардіографічні синдроми та феномени: звертаємося до підручника
 Редакція медичної газети «Здоров’я України», тематичного номера «Кардіологія. Ревматологія. Кардіохірургія» продовжує знайомити наших читачів із ґрунтовним і детальним підручником «Функціональна діагностика» за редакцією доктора медичних наук, професора О.Й. Жарінова, доктора медичних наук, професора Ю.А. Іваніва та кандидата медичних наук, доцента В.О. Куця. Пропонуємо вашій увазі розділ «Електрокардіографічні синдроми та феномени».
Редакція медичної газети «Здоров’я України», тематичного номера «Кардіологія. Ревматологія. Кардіохірургія» продовжує знайомити наших читачів із ґрунтовним і детальним підручником «Функціональна діагностика» за редакцією доктора медичних наук, професора О.Й. Жарінова, доктора медичних наук, професора Ю.А. Іваніва та кандидата медичних наук, доцента В.О. Куця. Пропонуємо вашій увазі розділ «Електрокардіографічні синдроми та феномени».
В електрокардіографії (ЕКГ) особливе місце належить діагностиці вроджених електричних хвороб («каналопатій»), асоційованих із підвищеним ризиком раптової серцевої смерті (РСС). Деякі автори об’єднують ці стани термінами «синдроми РСС», або «успадковані аритмії». Використання вказаних понять свідчить про єдність виявів фенотипу (злоякісні шлуночкові аритмії і РСС) та роль обтяженої спадковості у їхньому виникненні незважаючи на суттєві відмінності етіологічних факторів. Серед каналопатій, які можуть призвести до РСС, найбільше значення мають природжений синдром подовженого інтервалу QT, синдром укороченого інтервалу QT, синдром Бругада і катехоламінергічна поліморфна шлуночкова тахікардія (ШТ). На вказані хвороби, а також гіпертрофічну кардіоміопатію припадає більшість випадків РСС у підлітковому або молодому віці, зокрема у зв’язку із заняттями професійним спортом. Протягом останніх десятиліть з’явилося чимало нової інформації про ЕКГ-особливості каналопатій. Для лікаря функціональної діагностики важливо вміти розпізнавати ЕКГ-синдроми, асоційовані з підвищеним ризиком РСС, а також відрізняти їх від доброякісних феноменів, які можуть траплятися у здорових осіб. Наголосимо, що синдроми і феномени преекзитації шлуночків, що також можуть поєднуватися з підвищенням ризику життєво небезпечних аритмій серця, описано в попередньому розділі.
Причини та механізми виникнення РСС
В узгоджених рекомендаціях під раптовою смертю розуміють «нетравматичну неочікувану фатальну подію, яка трапляється протягом однієї години після початку симптомів у нібито здорової особи; якщо свідків події немає, визначення стосується випадків, коли в жертви не було ознак хвороби за 24 години до виникнення події». Окремо зазначено, що «РСС – це раптова смерть у пацієнта із природженою або набутою потенційно фатальною серцевою хворобою; або при автопсії виявлено серцеву чи судинну аномалію як причину події; або не виявлено очевидних екстракардіальних причин при посмертному дослідженні, що дає підстави думати про аритмію як імовірну причину смерті» (Priori S.G. et al., 2015). У багатьох випадках РСС є першим, але водночас фатальним виявом хвороби серця, і тому основним напрямом досліджень є пошук маркерів ризику і шляхів ефективної профілактики РСС.
РСС може виникати як при діагностованих раніше хворобах серця, так і в осіб, які до моменту смерті вважалися здоровими. На РСС припадає значна частка випадків смерті хворих на ішемічну хворобу серця або дилатаційну кардіоміопатію. В пацієнтів із безсимптомною дисфункцією ЛШ у структурі причин смерті домінує саме РСС. Але майже у 12% випадкiв причина РСС залишається незрозумiлою, оскільки при автопсiї або пiсля всебiчного обстеження пацiєнтiв, якi перенесли зупинку серця, не знаходять ознак структурного ураження серця. Вiдсоток хворих, якi помирають раптово без очевидного захворювання серця, є найбiльшим у молодому вiцi.
Основним безпосереднім механізмом РСС є шлуночкові тахіаритмії, переважно стійка мономорфна або (рідше) поліморфна ШТ із трансформацією у фібриляцію шлуночків (ФШ). Значно рідше (переважно у пацієнтів із тяжкими структурними ураженнями міокарда) до РСС призводять брадіаритмії, асистолія або електромеханічна дисоціація. Втім для здійснення стратифікації ризику та проведення профілактичних заходів важливо розуміти, що ШТ та/або ФШ можуть формуватися на тлі відмінних за своїм фенотипом патологічних станів. Їх найчастішою причиною є ішемічна хвороба серця або серцева недостатність, рідше спостерігаються природжені електричні та міокардіальні аритмогенні хвороби.
Синдром подовженого інтервалу QT
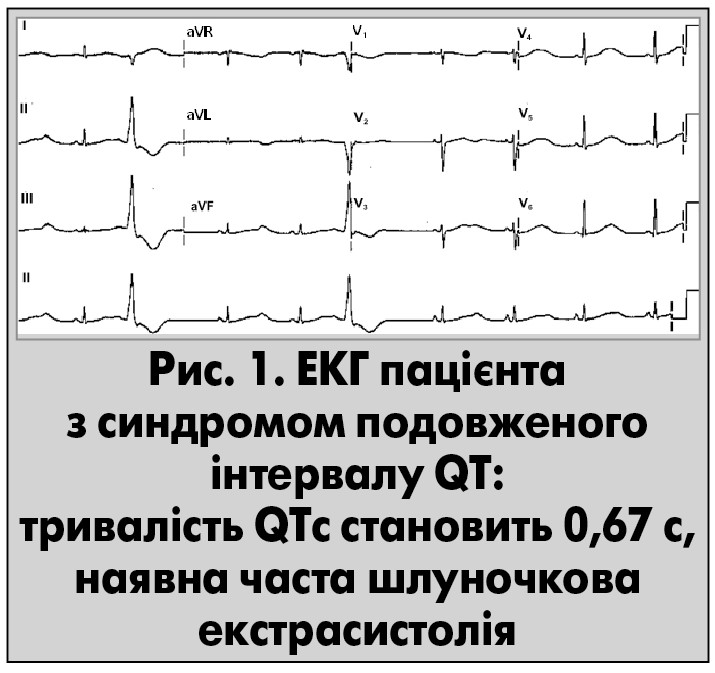 Особливою формою життєво небезпечних порушень серцевого ритму є полiморфнi ШТ (типу «пірует»), якi виникають у пацiєнтiв із природженим або набутим синдромом подовженого iнтервалу QT. Перше повiдомлення про вроджений синдром подовженого iнтервалу QT зробили A. Jervell i F. Lange-Nielsen 1957 р. У кiлькох членiв однiєї сiм’ї вони помiтили поєднання нейросенсорної глухонiмоти, подовження iнтервалу QT i РСС у молодому вiцi. У 1963 р. C. Romano, a в 1964 р. O. Ward повiдомили про автосомно-домiнантний характер успадковування подовження QT i РСС (без глухонімоти), на вiдмiну вiд автосомно-рецесивного типу синдрому Jervell – Lange-Nielsen. За різними даними, частота виявлення синдрому подовженого iнтервалу QT становить від 1 на 2500 до 1 на 10 тис. осіб (Gussak І., Antzelevitch C., 2008). Таким чином, якщо в Україні щороку народжується до півмільйона дітей, серед них може бути 50‑200 пацієнтів із синдромом подовженого iнтервалу QT і небезпекою РСС у молодому віці. Основними клінічними виявами є напади синкопе, судоми або РСС. При пірует-тахікардії традиційні підходи до антиаритмічної терапії і кардіоверсія малоефективні, а усунення провокативного чинника можливе лише за набутого синдрому подовженого iнтервалу QT.
Особливою формою життєво небезпечних порушень серцевого ритму є полiморфнi ШТ (типу «пірует»), якi виникають у пацiєнтiв із природженим або набутим синдромом подовженого iнтервалу QT. Перше повiдомлення про вроджений синдром подовженого iнтервалу QT зробили A. Jervell i F. Lange-Nielsen 1957 р. У кiлькох членiв однiєї сiм’ї вони помiтили поєднання нейросенсорної глухонiмоти, подовження iнтервалу QT i РСС у молодому вiцi. У 1963 р. C. Romano, a в 1964 р. O. Ward повiдомили про автосомно-домiнантний характер успадковування подовження QT i РСС (без глухонімоти), на вiдмiну вiд автосомно-рецесивного типу синдрому Jervell – Lange-Nielsen. За різними даними, частота виявлення синдрому подовженого iнтервалу QT становить від 1 на 2500 до 1 на 10 тис. осіб (Gussak І., Antzelevitch C., 2008). Таким чином, якщо в Україні щороку народжується до півмільйона дітей, серед них може бути 50‑200 пацієнтів із синдромом подовженого iнтервалу QT і небезпекою РСС у молодому віці. Основними клінічними виявами є напади синкопе, судоми або РСС. При пірует-тахікардії традиційні підходи до антиаритмічної терапії і кардіоверсія малоефективні, а усунення провокативного чинника можливе лише за набутого синдрому подовженого iнтервалу QT.
У середньому природжений синдром подовженого iнтервалу QT уперше виявляється у віці семи років, причому в однієї третини пацієнтів наявний сімейний анамнез. Вроджений синдром подовженого iнтервалу QT – це порушення реполяризації міокарда, яке виявляється тривалістю коригованого інтервалу QT більш ніж 440‑460 мс (табл. 1, рис. 1), морфологічними змінами зубця Т і підвищенням ризику виникнення пірует-тахікардії (Fisch Ch., Knoebel S.B., 2000). Виникнення такого синдрому пов’язують із мутаціями генів, які відповідають за функціонування калієвих, натрієвих або кальцієвих каналів у мембранах кардіоміоцитів. Описано принаймні 13 типів природженого синдрому подовженого iнтервалу QT: п’ять із них зумовлені змінами протеїнів, які є компонентами калієвих каналів, один – натрієвих, а один – протеїну плазматичної мембрани анкірину В.
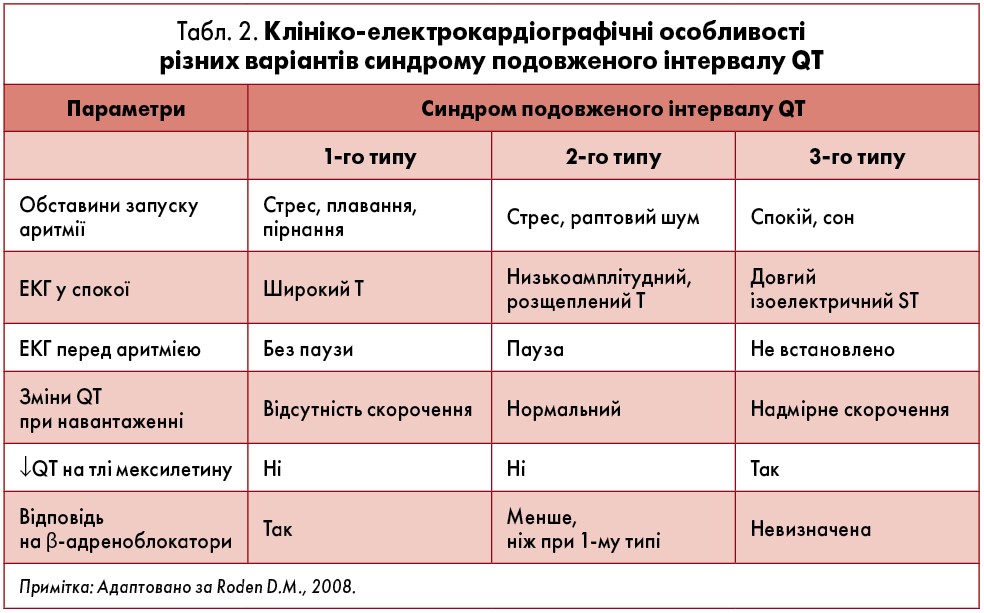
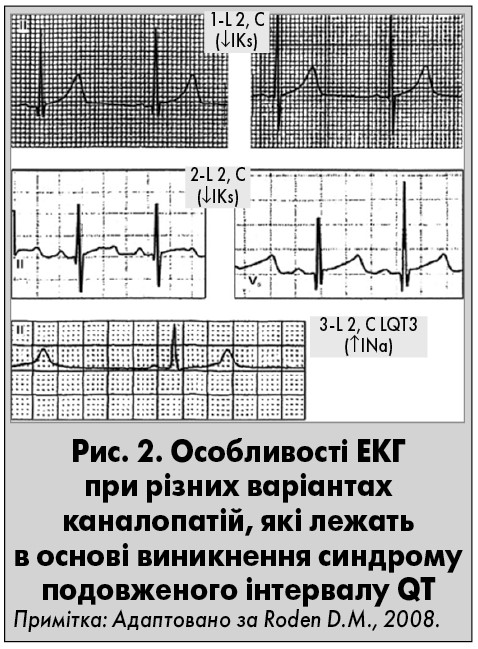 У пацiєнтiв із генетичними порушеннями, характерними для синдрому подовженого iнтервалу QT, гетерогенними є як механізми формування аритмій, так i вияви фенотипу. Зокрема, 6‑10% носiїв генетичних дефектiв взагалi не мають подовження iнтервалу QT, а десь у 6% пацiєнтiв пароксизми життєво небезпечних полiморфних ШТ або епiзоди синкопе виникають на тлі нормального iнтервалу QT у спокої (Zimetbaum P.J., Josephson M.E., 2009). Очевидно, провідним «пусковим» фактором нападів, що ведуть до РСС у осіб із синдромом подовженого iнтервалу QT, є раптова активація симпатоадреналової системи. Напади часто починаються після сильного фiзичного навантаження (зокрема плавання) чи психоемоцiйного збудження або, наприклад, пiсля того, як у повнiй тишi голосно вмикається будильник. Утім при окремих генетичних варіантах синдрому подовженого iнтервалу QT аритмії виникають у спокої або навіть під час сну (табл. 2). Крім того, різні генетичні варіанти синдрому можуть асоціюватися з різною графікою сегмента ST і зубця T (рис. 2).
У пацiєнтiв із генетичними порушеннями, характерними для синдрому подовженого iнтервалу QT, гетерогенними є як механізми формування аритмій, так i вияви фенотипу. Зокрема, 6‑10% носiїв генетичних дефектiв взагалi не мають подовження iнтервалу QT, а десь у 6% пацiєнтiв пароксизми життєво небезпечних полiморфних ШТ або епiзоди синкопе виникають на тлі нормального iнтервалу QT у спокої (Zimetbaum P.J., Josephson M.E., 2009). Очевидно, провідним «пусковим» фактором нападів, що ведуть до РСС у осіб із синдромом подовженого iнтервалу QT, є раптова активація симпатоадреналової системи. Напади часто починаються після сильного фiзичного навантаження (зокрема плавання) чи психоемоцiйного збудження або, наприклад, пiсля того, як у повнiй тишi голосно вмикається будильник. Утім при окремих генетичних варіантах синдрому подовженого iнтервалу QT аритмії виникають у спокої або навіть під час сну (табл. 2). Крім того, різні генетичні варіанти синдрому можуть асоціюватися з різною графікою сегмента ST і зубця T (рис. 2).
Ключові аспекти діагностики синдрому подовженого iнтервалу QT: ретельне дослідження обставин виникнення клінічних симптомів, оцінка сімейного анамнезу, а також аналіз ЕКГ. У нормі тривалість коригованого інтервалу QT, оціненого за формулою Базетта, є <0,43 с у чоловіків і <0,45 с у жінок (див. табл. 1).
Якщо клінічна картина дозволяє припускати аритмічне походження нападів синкопе і наявне подовження коригованого інтервалу QT >460‑500 мс, діагноз не викликає сумніву. Додаткове діагностичне значення можуть мати деякі інші ЕКГ-критерії: збільшення тривалості інтервалу від піку до моменту закінчення зубця Т >90 мс, великі негативні зубці Т/U, які передують злоякісним шлуночковим аритміям, синусова брадикардія. Наголосимо, що за відсутності характерних рис анамнезу і при нормальній тривалості інтервалу QT потреби у застосуванні додаткових методів діагностики і альтернативних ЕКГ-критеріїв для заперечення синдрому подовженого iнтервалу QT немає. В сумнівних випадках діагностичний пошук може передбачати навантажувальний тест з оцінкою змін тривалості інтервалу QT на тлі збільшення частоти серцевих скорочень, а також альтернації зубця Т. У розвинених країнах рутинно здійснюється генетичне тестування з метою виявлення найпоширеніших мутацій.
Оптимальне лікування синдрому подовженого iнтервалу QT слід призначати після генетичного аналізу. Виявлення природженого синдрому подовженого iнтервалу QT є підставою для заборони великих фізичних навантажень. Усім пацієнтам із таким синдромом, які перенесли зупинку серця і були успішно реанімовані, показано імплантацію автоматичного внутрішнього кардіовертера-дефібрилятора (АВКД). Очевидно, ці пристрої можуть бути рекомендовані також окремим пацієнтам із повторними епізодами синкопе або пірует-тахікардії, які не припиняються на тлі застосування β-адреноблокаторів, а також за наявності характерних клінічних симптомів у хворих із випадками РСС у молодому віці в родичів першого порядку.
Синдром укороченого інтервалу QT
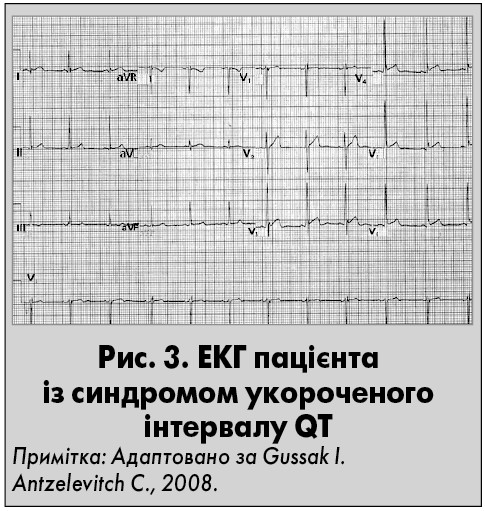 У 2000 р. українець І. Гусак, який наразі працює у США, разом зі своїми колегами описав синдром РСС у поєднанні з тривалістю інтервалу QT <300 мс (рис. 3). Хвороба вкрай небезпечна у всіх вікових групах, зокрема в дітей у перші місяці життя. Даних про поширеність такого синдрому поки що немає. При цьому вже відомо, що скорочення тривалості потенціалу дії та інтервалу QT асоціюється з певними генетичними мутаціями в п’яти генах. Окрім укорочення інтервалу QT, спостерігаються відсутність сегмента ST і симетричні високі зубці Т у грудних відведеннях.
У 2000 р. українець І. Гусак, який наразі працює у США, разом зі своїми колегами описав синдром РСС у поєднанні з тривалістю інтервалу QT <300 мс (рис. 3). Хвороба вкрай небезпечна у всіх вікових групах, зокрема в дітей у перші місяці життя. Даних про поширеність такого синдрому поки що немає. При цьому вже відомо, що скорочення тривалості потенціалу дії та інтервалу QT асоціюється з певними генетичними мутаціями в п’яти генах. Окрім укорочення інтервалу QT, спостерігаються відсутність сегмента ST і симетричні високі зубці Т у грудних відведеннях.
Для коректної діагностики цього синдрому важливо заперечити альтернативні причини зменшення тривалості QT, зокрема гіперкаліємію, гіпомагніємію, ацидоз, інтоксикацію серцевими глікозидами, застосування андрогенних анаболічних засобів тощо. Загалом синдром укороченого інтервалу QT діагностують за його тривалості ≤340 мс (Gussak І., Antzelevitch C., 2008). Слід також розглядати його наявність за тривалості QTс ≤360 мс та присутності одного або декількох таких станів, як-то підтверджена патогенна мутація, синдром укороченого інтервалу QT та РСС у віці до 40 років у сімейному анамнезі, перенесений епізод ШТ/ФШ з успішною реанімацією за відсутності структурної хвороби серця (Priori S.G. et al., 2015). Водночас у разі тривалості інтервалу QT <300 мс діагноз є безсумнівним.
Особам із синдромом укороченого інтервалу QT, в яких є симптоми небезпечних для життя аритмій серця, показано імплантацію АВКД. Частота рецидиву тяжкої аритмії становить 10% на рік. Пацієнтам, які відповідають вимогам для імплантації АВКД, але мають певні протипоказання або відмовляються від подібної процедури, може бути призначено терапію хінідином або соталолом, що, ймовірно, знизить частоту аритмічних подій (Priori S.G. et al., 2015). Доцільність застосування АВКД у безсимптомних осіб з обтяженим сімейним анамнезом на цей час залишається невизначеною.
Синдром Бругада
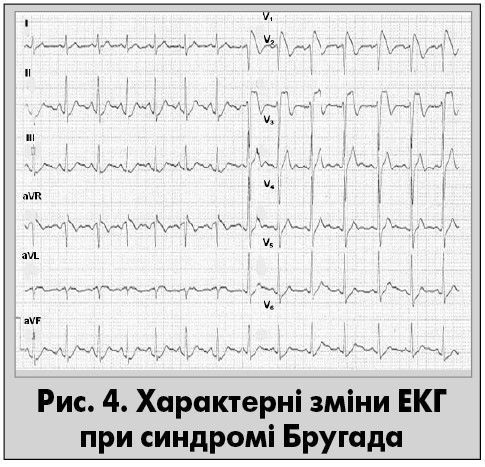 У 1992 р. брати Бругада описали новий синдром, який асоціюється з РСС у пацієнтів без структурної хвороби серця. Цей синдром характеризується особливим типом ЕКГ (псевдоблокада правої ніжки пучка Гіса, псевдоінфарктний пiдйом сегмента ST у відведеннях V1-V3, нерідко – атріовентрикулярна блокада І ступеня) сумісно з документованими епiзодами стійкої ШТ чи ФШ, або сімейним анамнезом РСС у молодому віці (рис. 4). Автори повiдомлення одразу запiдозрили, що ця хвороба має успадкований характер. Іонні каналопатії призводять до розладiв електричної функцiї серця i фатальних серцевих аритмiй. Окрім вказаних характерних рис, синдром Бругада може поєднуватися з нападами синкопе незрозумілого походження, пароксизмами поліморфної ШТ, які припиняються спонтанно, тяжкими порушеннями дихання уві сні (Gussak І., Antzelevitch C., 2008). При здійсненні інвазивного електрофізіологічного дослідження в таких пацієнтів нерідко індукується стійка ШТ. Наголосимо, що інколи зміни ЕКГ, характерні для синдрому Бругада, випадково виявляються в осіб без будь-яких клінічних ознак, характерних для нього (рис. 5). У таких випадках можна обмежитися динамічним спостереженням.
У 1992 р. брати Бругада описали новий синдром, який асоціюється з РСС у пацієнтів без структурної хвороби серця. Цей синдром характеризується особливим типом ЕКГ (псевдоблокада правої ніжки пучка Гіса, псевдоінфарктний пiдйом сегмента ST у відведеннях V1-V3, нерідко – атріовентрикулярна блокада І ступеня) сумісно з документованими епiзодами стійкої ШТ чи ФШ, або сімейним анамнезом РСС у молодому віці (рис. 4). Автори повiдомлення одразу запiдозрили, що ця хвороба має успадкований характер. Іонні каналопатії призводять до розладiв електричної функцiї серця i фатальних серцевих аритмiй. Окрім вказаних характерних рис, синдром Бругада може поєднуватися з нападами синкопе незрозумілого походження, пароксизмами поліморфної ШТ, які припиняються спонтанно, тяжкими порушеннями дихання уві сні (Gussak І., Antzelevitch C., 2008). При здійсненні інвазивного електрофізіологічного дослідження в таких пацієнтів нерідко індукується стійка ШТ. Наголосимо, що інколи зміни ЕКГ, характерні для синдрому Бругада, випадково виявляються в осіб без будь-яких клінічних ознак, характерних для нього (рис. 5). У таких випадках можна обмежитися динамічним спостереженням.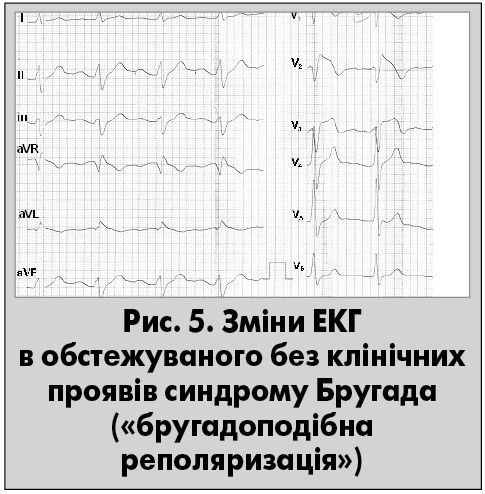
Синдром Бругада виявляють найчастіше в чоловiкiв, рідше – у жiнок і в дитячому вiцi. У дітей, в яких хворий один із батькiв, імовірність його виявлення становить 50%. Випадки цього синдрому були зареєстрованi у всьому свiтi, але особливо часто – в Японiї та Пiвденно-Східній Азiї. За даними братiв Бругада, РСС унаслiдок цього синдрому в Європi та Північній Америцi виникає приблизно в 1 на 10 тис. осіб на рiк. У середньому РСС стається у вiцi 40 рокiв. Події, зумовлені синдромом Бругада, переважно не пов’язані з фізичним навантаженням і можуть виникати під час відпочинку або сну. Провокативними чинниками є гарячка, надмірне споживання алкоголю та переїдання.
У пацієнтів із синдромом Бругада описані три варіанти змін сегмента ST і зубця Т (рис. 6). Ці зміни повинні реєструватися принаймні у двох правих грудних відведеннях. Класичний (перший) тип характеризується «псевдоінфарктною» графікою ЕКГ. Він виявляється низхідною елевацією сегмента ST принаймні на 2 мм з інверсією зубця Т (графіка «склепіння» – coved type). Частота виявлення такої графіки відрізняється у різних народностей: від 0,16% у деяких азійських популяціях до 1 на 3 тис. осіб в європейських країнах. При другому і третьому типах синдрому Бругада після початкового зниження сегмент ST переходить у позитивний зубець Т («сідловидна» графіка – saddleback type). Причому відмінність між цими типами визначається ступенем елевації сегмента ST: >1 мм для другого і <1 мм для третього. Втім при застосуванні фармакологічних проб із введенням антиаритмічних препаратів 1-го класу (аймалін, флекаїнід, прокаїнамід) можлива конверсія ЕКГ другого або третього типу в класичний тип синдрому Бругада. У випадках, коли є підозра на даний синдром, такі зміни ЕКГ у відповідь на фармакологічну пробу можуть мати ключове значення для підтвердження діагнозу.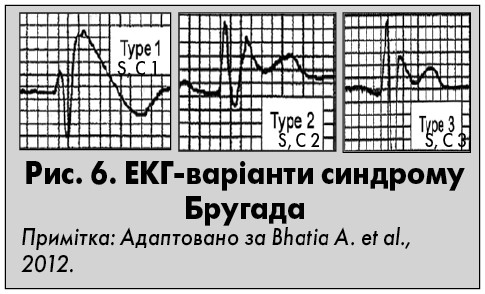
При розпiзнаннi синдрому Бругада здiйснюють скринiнгове обстеження всiх членiв сiм’ї. Обов’язково реєструють ЕКГ та здійснюють генетичний аналiз, а за необхiдностi – електрофізіологічне дослiдження. Єдиний ефективний метод лiкування – iмплантацiя АВКД, який розпiзнає аритмiю та негайно припиняє її електричним розрядом. Процедура рекомендована пацієнтам із синдромом Бругада, які вижили після зупинки серця або мають документовану спонтанну стійку ШТ, а також хворим з ЕКГ-картиною І типу та непритомністю в анамнезі (Priori S.G. et al., 2015). На жаль, жоден з антиаритмiчних препаратів не забезпечує адекватного захисту при цьому синдромi. Хінідин запропоновано як засіб для лікування «електричних штормів» у пацієнтів із синдромом Бругада, але немає даних, які підтвердили б його здатність знижувати ризик РСС (Priori S.G. et al., 2015). Якщо не імплантовано АВКД, 30% хворих помирають протягом трьох рокiв пiсля першого виникнення симптомiв.
Ідіопатична фібриляція шлуночків
Якщо у пацiєнта, реанімованого після зупинки серця внаслiдок ФШ, не виявлено жодних змін структурно-функцiонального стану міокарда та порушень ЕКГ, діагностують ідіопатичну ФШ. Подібно до інших каналопатій, перспектива досліджень її патогенезу пов’язана з виявленням генетичних маркерів РСС, а також структурних порушень на молекулярному рівні. У країнах Західної Європи і США з цією метою створено низку реєстрiв для узагальнення даних тривалого спостереження за пацiєнтами, реанiмованими пiсля iдiопатичної ФШ, яким iмплантували АВКД.
Катехоламінергічна поліморфна шлуночкова тахікардія
Сімейна катехоламінергічна поліморфна ШТ – вроджена хвороба, яка характеризується зв’язком з адренергічною активацією. Злоякісні шлуночкові аритмії виникають у періоди емоційного або фізичного стресу. Зазвичай у пацієнтів із катехоламінергічною поліморфною ШТ немає ознак структурних змін міокарда і порушень ЕКГ у спокої. Хвороба трапляється рідко, і повноцінних епідеміологічних даних не існує. Нерідко катехоламінергічна поліморфна ШТ виявляється у кількох членів однієї сім’ї. Генетичні дослідження дозволили встановити зв’язок цієї злоякісної аритмії з порушеннями захоплення кальцію кардіоміоцитами, зумовленими генними мутаціями. Основа діагностики – особистий або сімейний анамнез випадків синкопе або РСС на тлі фізичного чи психоемоційного навантаження. Важливим діагностичним засобом є проба з фізичним навантаженням, чутливість якої для виявлення катехоламінергічної поліморфної ШТ становить до 80%. Нерідко спостерігають двоспрямовані шлуночкові екстрасистоли, які переходять у пробіжки або пароксизми двоспрямованої ШТ (Bayes de Luna А., 2011).
Перебіг катехоламінергічної поліморфної ШТ злоякісний. Хвороба часто виявляється вже у дитячому віці, на відміну від синдрому подовженого iнтервалу QT і аритмогенної дисплазії правого шлуночка (ПШ), які можуть діагностуватися у підлітковому або дорослому віці. Важливе значення для її виявлення мають дані про наявність двоспрямованих шлуночкових екстрасистол при холтерівському моніторуванні ЕКГ. Інколи виявляють також епізоди брадіаритмій. До досягнення віку 40 років помирають близько 50% хворих, які не отримують лікування. Усім пацієнтам із катехоламінергічною поліморфною ШТ рекомендовано лікування β-адреноблокаторами (можливо у комбінації з флекаїнідом), а також уникати спортивних змагань, фізичних і психоемоційних навантажень (Priori S.G. et al., 2015). Якщо на тлі лікування β-адреноблокаторами продовжують виникати напади синкопе або індуковані навантаженням аритмії, можна розглянути доцільність імплантації АВКД.
Синдром ранньої реполяризації шлуночків
Реполяризація шлуночків на ЕКГ відображається сегментом ST, зубцями Т і U. Точка, де закінчується (повертається до ізолінії) зубець S і починається сегмент ST, називається точкою J. У 1951 р. R. Grant et al. описали зазубрину на низхідній частині шлуночкового комплексу, переважно у середніх грудних відведеннях (V2-V4). Автори назвали цю зазубрину «хвиля J» і пов’язали її з ранньою та швидкою реполяризацією шлуночків.
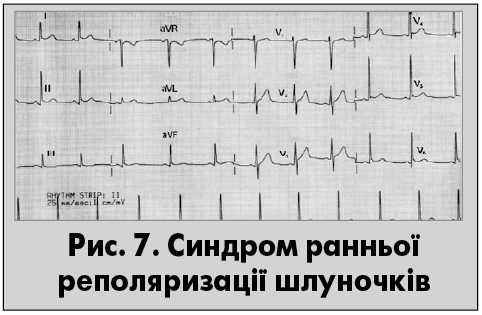 Рання реполяризація – це початок реполяризації в певній частині міокарда до закінчення деполяризації його інших ділянок. У пацієнтів із синдромом ранньої реполяризації шлуночків (СРРШ) виявляють підвищення точки J над ізолінією, елевацію сегмента ST із випуклістю донизу, високоамплітудні, гострі зубці Т, синусову брадикардію. Протягом тривалого часу СРРШ розглядався виключно як варіант норми (рис. 7). Утім зміни розташування точки J порівняно з ізолінією можна спостерігати при різноманітних станах, таких як гіпотермія (зубці Осборна), гіперкальціємія, ваготонія, ішемія міокарда, синдром Бругада. Останнім часом особливу увагу привертає можливий зв’язок окремих варіантів СРРШ із каналопатіями і РСС (Bastiaenen R., Behr E.R., 2012).
Рання реполяризація – це початок реполяризації в певній частині міокарда до закінчення деполяризації його інших ділянок. У пацієнтів із синдромом ранньої реполяризації шлуночків (СРРШ) виявляють підвищення точки J над ізолінією, елевацію сегмента ST із випуклістю донизу, високоамплітудні, гострі зубці Т, синусову брадикардію. Протягом тривалого часу СРРШ розглядався виключно як варіант норми (рис. 7). Утім зміни розташування точки J порівняно з ізолінією можна спостерігати при різноманітних станах, таких як гіпотермія (зубці Осборна), гіперкальціємія, ваготонія, ішемія міокарда, синдром Бругада. Останнім часом особливу увагу привертає можливий зв’язок окремих варіантів СРРШ із каналопатіями і РСС (Bastiaenen R., Behr E.R., 2012).
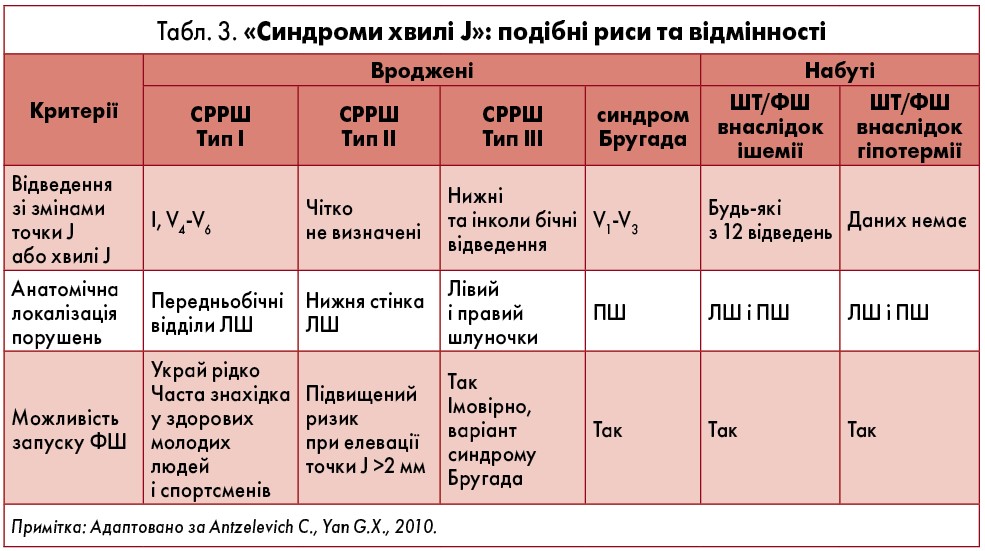
У загальній популяції поширеність СРРШ (під яким розуміють елевацію точки J у двох послідовних відведеннях принаймні на 0,1 мВ) оцінюють у 2‑5%. Переважно СРРШ спостерігають у молодих осіб – частіше чоловіків, які займаються спортом. Використання суворіших критеріїв (підвищення точки J >0,2 мВ, наявність ознак СРРШ не лише у лівих грудних, а й у нижніх відведеннях) зменшує поширеність СРРШ до 0,3‑0,8%. Можливе підвищення ризику РСС, зокрема внаслідок ідіопатичної ФШ, асоціюють із виразною елевацією точки J і сегмента ST у нижніх та/або лівих грудних відведеннях. Різні варіанти СРРШ пропонують позначати як «синдроми хвилі J» (Antzelevich C., Yan G.X., 2010) (табл. 3). Вказаний аспект залишається предметом клініко-епідеміологічних і генетичних досліджень.
Виявлення наявності каналопатій є важливим завданням обстеження молодих пацієнтів із нападами серцебиття, зареєстрованими шлуночковими аритміями або синкопальними станами. Початок діагностичного пошуку полягає в ретельному аналізі скарг, зокрема обставин виникнення нападів синкопе. У пацієнтів із високою імовірністю небезпечних для життя шлуночкових аритмій показане застосування додаткових методів візуалізації серця (ехокардіографії, за необхідності – магнітно-резонансної візуалізації), навантажувального тесту, в окремих випадках – електрофізіологічного дослідження. Другий важливий напрям – детальний сімейний анамнез, особливо за наявності у близьких родичів випадків РСС у молодому віці. Третій елемент загального обстеження – пошук на ЕКГ ознак, які асоціюються з підвищеним ризиком РСС (подовженого або вкороченого коригованого інтервалу QT, змін комплексу QRS, характерних для синдрому Бругада та інших).
Серед засобів первинної та вторинної профілактики РСС найбільш ефективним є імплантація АВКД. В окремих ситуаціях корисним може бути додаткове застосування антиаритмічних препаратів, передусім аміодарону, соталолу і β-адреноблокаторів. Перспективи верифікації діагнозу успадкованих аритмічних синдромів значною мірою залежать від доступності сучасних медичних технологій, зокрема, можливості проведення генетичних досліджень.
Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» № 4 (65) вересень 2019 р.