


30 жовтня, 2019
Фармакогенетика метформіну
Метформін – беззаперечний лідер серед препаратів для лікування цукрового діабету (ЦД) 2 типу. Нині метформін включений до всіх алгоритмів із ведення ЦД 2 типу, виданих професійними товариствами ендокринологів, як препарат першої лінії, що має бути призначений пацієнту одразу після встановлення цього діагнозу [1-3] за умови відсутності відповідних протипоказань (приміром, тяжка ниркова або печінкова недостатність). Це пов’язано з тим, що з доступних сьогодні засобів різних класів для лікування ЦД 2 типу саме метформін поєднує в собі всі 3 кардинальні переваги оптимального протидіабетичного препарату: він є безпечним, ефективним і дешевим [4, 5]. Терапія метформіном добре переноситься [6, 7]. Загалом метформін характеризується високою ефективністю, це один із небагатьох пероральних цукрознижувальних засобів, які сприяють зменшенню маси тіла, а не її збільшенню [6, 8, 9]. Зумовлене прийомом метформіну зниження ваги є стійким та залежить від прихильності до лікування [11]. Наразі метформін – один із найбільш широко вживаних препаратів для лікування ЦД 2 типу в усьому світі: так, у 2010 р. лише в США на цей препарат було виписано понад 48 млн рецептів.
Незважаючи на всі ці сприятливі властивості, метформін, на жаль, не можна вважати панацеєю. Клінічна практика й опубліковані дослідження свідчать, що монотерапії метформіном далеко не завжди достатньо для досягнення адекватного глікемічного контролю. Дуже часто виникає потреба в інтенсифікації лікування [15]; приблизно 21% пацієнтів, що розпочали прийом метформіну, не досягають цільових рівнів глікемії в перші 5 років терапії [16]. Довготривале спостереження в рамках Програми профілактики діабету (Diabetes Prevention Program, DPP) продемонструвало, що в учасників, рандомізованих у групу профілактичного прийому метформіну, з часом усе одно розвивався ЦД 2 типу – навіть під час лікування і незважаючи на доволі високий рівень прихильності до терапії (майже 80%). Ці дані свідчать, що метформін може лише затримувати (у середньому приблизно на 12 міс) дебют ЦД 2 типу, а не запобігати розвитку захворювання [9].
Отже, описаний сценарій із метформіном ілюструє ключовий бар’єр у сучасній фармакотерапії: фокусуючись на «середньостатистичному пацієнті» (який типово являє собою образ учасника успішного клінічного дослідження та «ідеально» відповідає на лікування), ми нехтуємо охопленням усього спектра можливих відповідей на терапію – і через це не вдається виявити тих пацієнтів, у яких певне лікування може виявитися недоцільним. У разі ЦД 2 типу значний час може бути витрачений на спроби досягти глікемічного контролю за допомогою стандартної монотерапії метформіном у тих пацієнтів, у яких це aпріорі неможливо. Для того щоб на практиці реалізувати концепцію «персоналізованого лікування», необхідно відійти від стереотипних уявлень про «середньостатистичних» пацієнтів та почати виділяти окрему підгрупу хворих, у яких може бути доцільним починати лікування одразу з більш високої дози метформіну або взагалі з альтернативного цукрознижувального препарату.
Однак слід визнати, що наразі лікарі вкрай обмежені у своїй здатності визначати тих пацієнтів, які не дадуть адекватної відповіді на терапію метформіном. Частково це пов’язано з недостатністю глибинних знань щодо специфічної молекулярної мішені, на яку впливає метформін. Відсутність цих даних, з одного боку, перешкоджає розробці дизайну експериментів, які мали б на меті описати групу пацієнтів, що, ймовірно, не будуть відповідати на лікування, але водночас не виключає можливості проведення агностичних генетичних скринінгів, результатом яких цілком можуть стати фундаментальні наукові відкриття.
Генетичний підхід як потенційне вирішення проблеми
Деякі індивідуальні відмінності, що лежать в основі варіабельності відповіді на терапію метформіном, імовірно, мають генетичний характер. Наприклад, епідеміологічні дослідження свідчать про існування суттєвих відмінностей у відповіді на метформін у пацієнтів різних етнічних груп [19] – в основі деяких із них вочевидь лежить саме генетичний компонент. Нещодавно дослідникам став доступний метод аналізу всього геному, який дає можливість з адекватною точністю оцінити внесок генетичних факторів (зокрема, однонуклеотидних поліморфізмів, ОНП) у варіабельність відповіді на лікування метформіном [20]. Це – повногеномний пошук асоціацій (GWAS, Genome-Wide Association Studies), що являє собою напрям генетичних досліджень, пов’язаних із вивченням асоціацій між геномними варіантами та певними фенотиповими ознаками. Для оцінки відповіді на метформін у пацієнтів із ЦД 2 типу метод GWAS був уперше застосований у ході дослідження GoDARTS (Genetics of Diabetes and Audit Research Tayside Study) [21], до якого було включено більш як 17 тис. пацієнтів із діагностованим ЦД 2 типу; їхні дані зіставляли з даними учасників контрольної групи без ЦД 2 типу. Застовуючи повногеномний метод аналізу складних ознак [22], Zhou і співавт. проаналізували доступні дані 2085 учасників та дійшли висновку, що внесок спадкового фактора у визначення відповіді на метформін варіює від 20 до 34% (залежно від специфічного оцінюваного критерію відповіді на терапію) [23].
До розробки методу GWAS, створеного для детального вивчення геному в цілому, дослідження метформіну були зосереджені на клітинних транспортерах, які контролюють його метаболізм. З появою методу GWAS ситуація кардинально змінилася, і в наступних розділах статті в хронологічному порядку описані результати цих сучасних фармакогенетичних досліджень.
Перша фаза: дослідження генів-кандидатів
Оскільки метформін є гідрофільною молекулою, він не може легко проникати крізь клітинні мембрани. Натомість він активно транспортується за допомогою білків – транспортерів органічних катіонів в ентероцити, гепатоцити та ниркові епітеліальні клітини (рис.).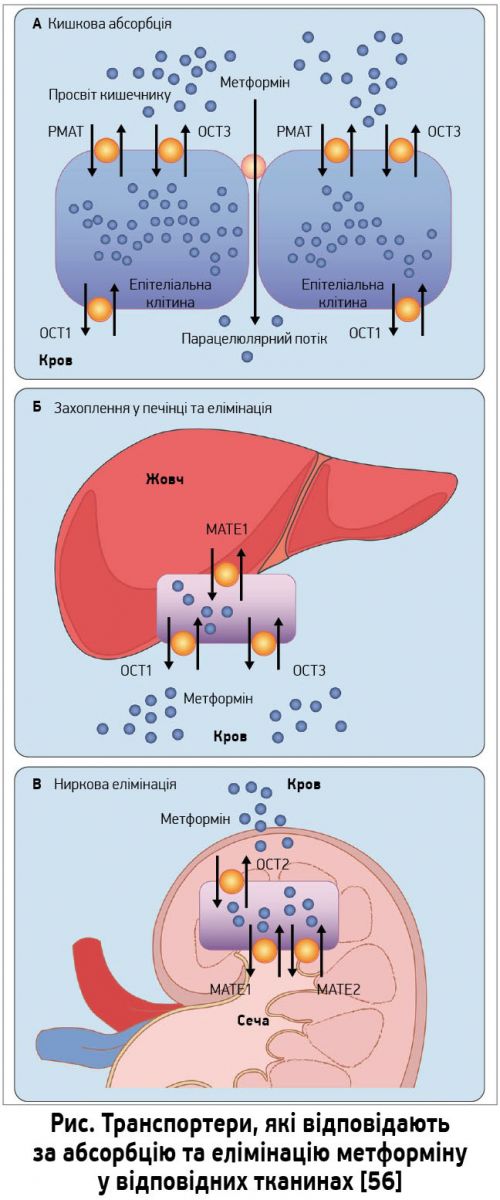
Після перорального прийому метформін досягає ендотелію кишечнику (А), де він захоплюється ентероцитами за допомогою плазмового транспортера моноамінів (PMAT; кодується геном SLC29A4) та транспортера органічних катіонів (OCT3; кодується геном SCL22A3) – вони обидва локалізовані на люмінальній поверхні кишкового епітелію [26, 27]. OCT1 транспортує метформін з ентероциту до серозного боку ендотелію. Коли метформін досягає печінки (Б), OCT1 та OCT3 сприяють його захопленню гепатоцитами [25, 27-29]. Виведення метформіну в жовч здійснюється за допомогою білка екструзії лікарських засобів і токсинів (MATE1; кодується геном SLC47A1) [31, 32]; він захоплюється епітеліальними клітинами в нирках (В) за допомогою OCT2 (кодується геном SLC22A2), у той час як MATE1 та MATE2 (останній кодується геном SLC47A2) беруть участь в екскреції незміненого метформіну із сечею [31, 32, 34].
Найбільш вивченим білком-транспортером є OCT1. Ген, що кодує OCT1 – SLC22A1, є високополіморфним та має низку місенс-мутацій ОНП, які впливають на його активність [36-39]. Група дослідників під керівництвом К. Giacomini вперше продемонструвала, що наявність як мінімум одного з 4 варіантів кодування зниженої функції цього транспортера (R61C/rs12208357, G401S/rs34130495, M420del/rs72552763 та/або G465R/rs34059508) послаблює ефекти короткого курсу терапії метформіном [30] – імовірно, шляхом порушення внутрішньоклітинного транспорту препарату [40]. Однак під час великого ретроспективного обсерваційного клінічного когортного дослідження за участю 1531 особи (дані про них отримали в ході дослідження GoDARTS) було встановлено, що 2 найчастіші варіанти ОНП, що кодують знижену функцію цього транспортера (R61C та M420del), не були асоційовані з 4 різними критеріями глікемічної відповіді на метформін (а саме з початковим рівнем зниження глікозильованого гемоглобіну HbA1c, ймовірністю досягнення цільового рівня HbA1c <7%, середнім рівнем HbA1c на тлі монотерапії метформіном або ризиком неефективності монотерапії, що визначався як необхідність призначення другого цукрознижувального препарату) [41].
У проспективному дослідженні South Danish Diabetes Study за участю 105 осіб [42] як залишкові рівноважні концентрації метформіну, так і зміна рівня HbA1c через 6 міс були меншими при збільшенні кількості алелів, що кодують зниження функції, – R61C, G401S, M420del та G465R [43]. Більш свіже дослідження, де використовувалися радіоізотопні мітки, продемонструвало, що накопичення метформіну в людських гепатоцитах зменшується у носіїв варіантів M420del та R61C гена SLC22A1 (без змін циркулюючих рівнів препарату) [44].Дослідження побічних ефектів, виконане дослідниками GoDARTS у 251 учасника з непереносимістю метформіну та в 1915 осіб, які добре переносили метформін, показали, що наявність ≥2 алелів, що кодують знижену функцію транспортера (R61C, C88R/rs55918055, G401S, M420del або G465R), збільшує ризик непереносимості метформіну більш як удвічі – переважно шляхом індукування накопичення метформіну в ентероцитах [45]; на тлі супутнього застосування
Друга фаза: GWAS
Співробітництво між дослідниками груп GoDARTS та UKPDS (UK Prospective Diabetes Study) призвело до першого застосування методу GWAS для оцінки відповіді на метформін [21]. У помірній за розміром виборці з 1024 учасників GoDARTS сигнал про асоціацію з відповіддю на метформін (визначеною як досягнення клінічного цільового значення HbA1c <7% протягом 18 міс з початку лікування або як кількісна зміна рівня HbA1c) був виявлений навколо гена, що кодує мутантну кіназу при атаксії-телеангіектазії (ATM). Найсильніша асоціація (ймовірність досягнення цільового рівня глікемії, 1,6) була виявлена на рівні ОНП rs11212617; цей сигнал був прослідкований у незалежній виборці з 1783 учасників дослідження GoDARTS та 1113 учасників дослідження UKPDS, досягнувши статистичної достовірності по всьому геному. Інші групи дослідників встановили відтворюваність цього результату в учасників із діагностованим ЦД 2 типу [51].
У метааналізі MetGen Consortium (n=10 557) повногеномна статистично достовірна асоціація з відповіддю на метформін спостерігалася на рівні ОНП rs8192675 – інтрону білка – транспортера глюкози GLUT2, що кодується геном SLC2A2 та експресується в гепатоцитах [54]. Білок – транспортер GLUT2 переносить глюкозу крізь клітинну мембрану шляхом полегшеної дифузії. Вважається, що зменшення продукції глюкози в печінці, досягнуте на тлі прийому метформіну, опосередковане саме цим транспортером. Цікаво, що той самий алель, який був асоційований із покращенням відповіді на метформін (тобто з нижчими значеннями HbA1c на тлі лікування),також асоціювався з більш високим вихідним рівнем HbA1c, це узгоджується з попередніми повідомленнями про асоціацію іншого ОНП у цьому локусі з рівнем глюкози натщесерце в осіб без ЦД [55]. Рівні експресії GLUT2 в печінці також були достовірно асоційовані з тим самим ОНП (алель, пов’язаний із вищим вихідним рівнем HbA1c, асоціювався з меншою експресією SLC2A2).
Висновки та майбутні напрями
Для того щоби забезпечити цілеспрямоване застосування метформіну в найбільш оптимальній популяції пацієнтів із ЦД 2 типу, необхідно поглибити розуміння молекулярного механізму дії препарату. Масштабні генетичні дослідження за участю пацієнтів, що приймають метформін, уже виявили певні генетичні особливості, які в перспективі зможуть пояснити, чому одні пацієнти реагують на нього краще, а інші – гірше. На практиці виявлення та підтвердження нових потужних фармакогенетичних асоціацій може стати основою для категоризації генетичного ризику, який значною мірою пояснить варіабельність відповіді на метформін або ймовірну появу побічних ефектів. На основі таких знань можна буде створювати автоматизовані алгоритми прийняття рішень, які дадуть можливість лікарям точно розраховувати необхідну дозу метформіну як препарату першої лінії або ж у деяких випадках одразу призначати альтернативні цукрознижувальні препарати.
Стаття друкується в скороченні.
Список літератури знаходиться в редакції.
Diabetologia (2017); 60: 1648-1655. DOI: 10.1007/s00125-017-4335-y.
Переклала з англ. Олена Зотова
Медична газета «Здоров’я України 21 сторіччя» № 19 (464), жовтень 2019 р.