


23 грудня, 2019
Мукополісахаридози: очні прояви
 За даними світової літератури, близько 5% новонароджених з’являються на світ із тією чи іншою генетичною патологією, яка в 60% випадків скорочує тривалість життя і в 70% – зменшує репродуктивні можливості [1]. Спадкові хвороби органа зору є причиною сліпоти приблизно в 42-84% випадків і, незважаючи на невелику поширеність, посідають важливе місце в ланці охорони здоров’я, оскільки є основним чинником інвалідизації дитячого населення [2].
За даними світової літератури, близько 5% новонароджених з’являються на світ із тією чи іншою генетичною патологією, яка в 60% випадків скорочує тривалість життя і в 70% – зменшує репродуктивні можливості [1]. Спадкові хвороби органа зору є причиною сліпоти приблизно в 42-84% випадків і, незважаючи на невелику поширеність, посідають важливе місце в ланці охорони здоров’я, оскільки є основним чинником інвалідизації дитячого населення [2].
Медична генетика, що вивчає етіологію, патогенез, клінічні прояви спадкової патології та забезпечує розробку методів профілактики і лікування спадкових захворювань, та медико-генетичне консультування є важливими складовими сучасної офтальмології, адже саме вони в подальшому мають забезпечити покращення якості життя маленьких пацієнтів. Основними напрямами офтальмогенетики є вивчення генетично зумовленої патології органа зору – як ізольованої, так і в складі синдромів; визначення тактики адекватного комплексного терапевтичного та хірургічного лікування генетичної патології органа зору; визначення можливого методу генетичного прогнозування, виявлення членів сімей із доклінічною стадією захворювання; формування, ведення реєстру сімей зі спадковими очними захворюваннями та їх спостереження; розробка науково-практичних програм, інформаційного забезпечення, рекомендацій щодо ранньої діагностики та профілактики, патогенетично орієнтованої терапії та можливого хірургічного лікування спадкових захворювань органа зору; підготовка сімейних лікарів, офтальмологів, медичних генетиків і підвищення їх кваліфікації [2].
Серед генетичних захворювань, при яких уражається орган зору, важливе місце належить хворобам накопичення. На сьогодні зареєстровано понад 40 таких захворювань. Особливе значення з-поміж них мають мукополісахаридози (МПС), оскільки майже при кожному з 9 типів МПС зустрічаються ті чи інші зміни з боку ока.
Глобальна поширеність МПС становить 1 випадок на 162 тис. новонароджених. За даними національних общин хворих на МПС, усього у світі на цю патологію страждає близько 1500 людей. Згідно з епідеміологічними дослідженнями, захворюваність на МПС у різних країнах варіює від 1:15 000 (у Чехії) до 1:156 000 (у Німеччині). Так, за даними літератури, в Австрії цей показник дорівнює 1:78 000 новонароджених; у Франції – 1:80 000; в Австралії – 1:136 000; у Нідерландах – 1:149 000, у Великій Британії – 1:150 000 [18]. На жаль, у доступних літературних джерелах даних щодо захворюваності на МПС в Україні немає.
МПС уперше були описані C. Hunter у 1917 р. Автор спостерігав двох братів віком 8 і 10 років з порушенням опорно-рухового апарату, гепато-, сплено- і кардіомегалією, а також зі зниженням інтелекту. У 1919 р. G. Gurler описала ідентичну, але більш тяжку клінічну картину захворювання двох хлопчиків, які не були родичами. Ця патологія увійшла в літературу під назвою «мукополісахаридози» у 1952 р. завдяки дослідженням G. Brante, який виділив у печінці хворих фракцію, що містила гексозамін й уронову кислоту. У подальшому було встановлено, що ці речовини є глікозаміногліканами.
МПС – група спадкових хвороб сполучної тканини, спричинених порушенням обміну глікозаміногліканів (кислих мукополісахаридів) у результаті генетично зумовленої неповноцінності ферментів, що беруть участь у їх розщепленні. Успадковується хвороба за аутосомно-домінантним типом. При МПС уражається система лізосомальних ферментів, що беруть участь у катаболізмі глікозаміногліканів. Таким чином, у результаті ферментативної недостатності останні накопичуються в органах і тканинах.
Глікозаміноглікани можна виявити в багатьох органах і тканинах. Так, хондроїтинсульфат і кератансульфат є компонентами хрящової, кісткової тканини, а також тканин ока; дерматансульфат типовий для шкіри та клапанів серця; гепарансульфат є компонентом мембран нервових клітин і тканин мозку. Моніторинг накопичення глікозаміногліканів дозволяє встановити і відслідкувати клінічні прояви МПС.
За сучасною класифікацією, виокремлюють 9 типів МПС, кожен з яких зумовлений дефіцитом специфічного ферменту, що бере участь у послідовному розщепленні певного глікозаміноглікану (хондроїтин-, дерматан-, гепаран- і кератансульфатів):
- МПС типу I (синдром Гурлер) – дефект ферменту альфа-L‑ідуронідази;
- МПС типу I (синдром Гурлер-Шейе) – дефект ферменту альфа-L‑ідуронідази;
- МПС типу I (синдром Шейе) – дефект ферменту альфа-L‑ідуронідази;
- МПС типу IІ (синдром Хантера) – дефект ферменту ідуронат‑2-сульфатази;
- МПС типу IІІ (синдром Санфіліппо) – дефект 4 ензимів;
- МПС типу IV (синдром Моркіо) – дефект ферменту N‑ацетил-галактозамін‑6-сульфатази;
- МПС типу VI (синдром Марото-Ламі) – дефект ферменту арилсульфатази В;
- МПС типу VIІ (синдром Слая) – дефект ферменту бета-глюкуронідази;
- МПС типу ІX – дефект ферменту гіалуронідази [3].
Діагностика МПС є важливою на ранніх етапах захворювання, оскільки дає можливість своєчасно розпочати замісне лікування й адекватну симптоматичну терапію, а отже, продовжити повноцінне життя пацієнтів. У багатьох хворих МПС тривалий час залишається недіагностованим, що призводить до поступового розвитку необоротних патологічних змін в організмі, а в подальшому – до передчасної смерті. Більшість таких дітей при народженні не відрізняються від здорових, але в міру прогресування захворювання мультисистемні ураження даються взнаки. Найчастіше уражаються нервова, серцево-судинна, гепато-лієнальна системи, кістково-хрящова тканина та орган зору.
Причиною пізньої діагностики МПС найчастіше є значне різноманіття проявів патології та низька обізнаність медичних працівників. Пацієнти можуть тривалий час спостерігатись у ревматолога, невролога, кардіолога, залишаючись без остаточного діагнозу [4]. Зміни органа зору в таких пацієнтів найчастіше спостерігаються вже на початку захворювання в результаті накопичення глікозаміногліканів у тканинах ока.
За даними літератури, мембранозв’язувальні вакуолі, в яких розміщені глікозаміноглікани, були знайдені практично в усіх тканинах ока, що й забезпечило значну кількість очних проявів [5]. З‑поміж останніх до основних відносяться помутніння рогівки, офтальмогіпертензія, глаукома, дистрофія сітківки, атрофія та набряк зорового нерва (табл. 1.) [5, 6]. Крім того, може спостерігатися прогресування псевдоекзофтальму через мілку орбіту, гіпертелоризм, косоокість, гіперметропію й астигматизм [7].
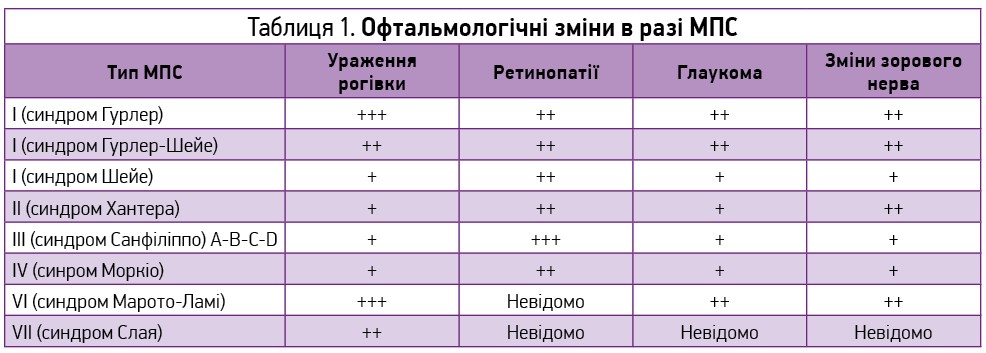
Розглянемо основні очні прояви МПС. Помутніння рогівки найчастіше виникає при МПС типів І, VI, VII [5], найбільш виражене в пацієнтів із МПС типу VI (рис. 1). Причина – структурні зміни кератоцитів і колагенових волокон строми рогівки через накопичення в них глікозаміногліканів внутрішньо та позаклітинно [7, 8]. На ранніх етапах помутніння рогівки може мати безсимптомний перебіг або ж хворі можуть скаржитися на підвищену світлобоязнь [9]. Цей стан найчастіше має тенденцію до прогресування з поступовим погіршенням гостроти зору, що може призвести до слабкозорості чи сліпоти, особливо при помутнінні в оптичній зоні рогівки [10].
Внутрішньоочна гіпертензія та глаукома в основному виявляються в пацієнтів із МПС типів І та VІ. Найчастіше виникають вторинно в результаті відносного звуження кута передньої камери чи погіршенні відтоку через обструкцію трабекулярної системи [11]. Клінічні прояви є характерними відповідно до типу глаукоми і характеризуються розширенням екскавації, підвищенням внутрішньоочного тиску та звуженням поля зору. У разі пізньої діагностики необоротно призводять до сліпоти.
Дегенерація сітківки частіше спостерігається при МПС типів І-ІV. Причиною її розвитку є відкладання глікозаміногліканів у пігментному епітелії сітківки та фоторецепторах, що в подальшому призводить до прогресуючої втрати останніх, розвитку дегенеративних змін сітківки та атрофії зорового нерва [12]. На ранніх етапах пацієнти можуть скаржитися на погіршення зору в темряві (гемералопія) [5]. Потім виникають звуження полів зору до трубчастого та втрата центрального зору. Дегенерація сітківки прогресує повільно, при цьому час початку патологічних змін залежить від тяжкості фенотипу [13].
Набряк диска зорового нерва й атрофія зорового нерва найчастіше мають місце при МПС типів І і VІ [12]. Унаслідок відкладання глікозаміногліканів у твердій мозковій оболонці та склері спостерігається потовщення субарахноїдального простору, що призводить до стискання зорового нерва та в подальшому сприяє розвитку його набряку і вторинної атрофії [12, 14, 20]. Набряк диска зорового нерва може також виникнути через підвищення внутрішньочерепного тиску [14]. Можливий й інший варіант розвитку атрофії, що виникає при безпосередньому відкладанні глікозаміногліканів у гангліозних клітинах і призводить до дегенерації нейронів. Атрофія зорового нерва спричиняє зниження контрастної чутливості, звуження полів зору, а в перспективі – втрату зору.
Офтальмологічні прояви досить часто виникають на ранніх етапах розвитку МПС [12]. Такі симптоми, як світлобоязнь, куряча сліпота (гемералопія), помутніння рогівки, псевдоекзофтальм, косоокість і зниження зору, досить легко можуть бути виявлені лікарями загальної практики шляхом огляду, опитування пацієнта чи його батьків. Тож за наявності нетипового зовнішнього вигляду і вище перерахованих симптомів лікар загальної практики має направити пацієнта з метою подальшого обстеження до генетика, педіатра й офтальмолога (табл. 2).
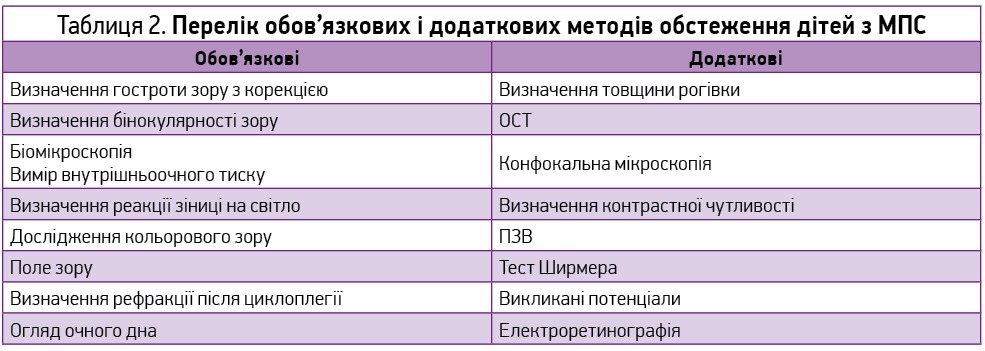
Необхідно відмітити, що обстеження таких пацієнтів може бути ускладнене через соціальні причини, як-от розлади інтелекту, що погіршує контакт з пацієнтом; небажання останнього співпрацювати тощо. На якість діагностики захворювання, безумовно, впливають і медичні причини. Так, при ураженні рогівки огляд може ускладнюватися світлобоязню [15]; помутніння рогівки може погіршувати візуалізацію очного дна, що ускладнює моніторинг набряку зорового нерва, атрофії зорового нерва, дегенерації сітківки; тяжкі помутніння рогівки та її потовщення можуть перешкоджати коректній оцінці кута передньої камери та внутрішньоочного тиску; також існують труднощі диференційної діагностики змін поля зору, пов’язаних із глаукомою та дегенеративними змінами сітківки [5, 16, 19]. Усе це необхідно враховувати при огляді пацієнтів.
Висновки
Скринінг і рання діагностика МПС є надзвичайно важливими з погляду профілактики інвалідності. Зважаючи на частоту офтальмологічних проявів МПС, необхідно вдосконалити програму підготовки спеціалістів охорони здоров’я загальної мережі (дільничних терапевтів, лікарів загальної практики – сімейної медицини) та спеціалізованих закладів охорони здоров’я (офтальмологічної служби, лікарів-спеціалістів) з питань скринінгу, діагностики і лікування МПС, диспансеризації та реабілітації хворих на МПС.
Список літератури знаходиться в редакції.
Медична газета «Здоров’я України 21 сторіччя» № 22 (467), листопад 2019 р.