


15 квітня, 2020
Хронічна лімфоцитарна лейкемія: від розпачу до надії
 Усі злоякісні гематологічні захворювання можна розподілити на лейкози (лейкемії) та лімфоми, а хронічна лімфоцитарна лейкемія (ХЛЛ) є найчастішою формою лейкемії.
Усі злоякісні гематологічні захворювання можна розподілити на лейкози (лейкемії) та лімфоми, а хронічна лімфоцитарна лейкемія (ХЛЛ) є найчастішою формою лейкемії.
Вважається, що лейкоз існував завжди. Ймовірно, першим пацієнтом з ХЛЛ, випадок якого описав у 1827 р. лікар Armand Velpeau, був 63-річний парижанин Верніс. Чи був описаний випадок першим повідомленням про ХЛЛ? Точної відповіді ми не дізнаємось, але історія ідентифікації цього захворювання налічує вже майже 200 років [1].
За оцінкою Американського товариства раку, у 2020 р. у США буде зафіксовано близько 60 530 нових випадків захворювання на усі види лейкозу, зокрема приблизно 21 040 нових випадків захворювання на ХЛЛ, а також близько 23 100 смертей від цих хвороб, у тому числі майже 4060 випадків смерті від ХЛЛ.
 У країнах з розвинутою економікою, у яких належно збираються епідеміологічні дані, рівень захворюваності та поширеності ХЛЛ досить рівномірний. Так, наприклад, у США від 1/4 до 1/3 усіх нових випадків лейкозів складає ХЛЛ. У пересічної людини протягом життя ризик захворіти на ХЛЛ становить приблизно 1:175 (0,57%). Цей ризик дещо вищий у чоловіків, ніж у жінок (1,3:1). ХЛЛ уражає в основному дорослих, середній вік на момент встановлення діагнозу становить близько 70 років. Хвороба рідко спостерігається в осіб до 40 років і надзвичайно рідко зустрічається у дітей [2]. ХЛЛ характеризується расовою схильністю: найчастіше зустрічається у представників європеоїдної раси, а найрідше – у монголоїдної (північно-східний вектор зменшення захворюваності), захворюваність у афроамериканців є проміжною.
У країнах з розвинутою економікою, у яких належно збираються епідеміологічні дані, рівень захворюваності та поширеності ХЛЛ досить рівномірний. Так, наприклад, у США від 1/4 до 1/3 усіх нових випадків лейкозів складає ХЛЛ. У пересічної людини протягом життя ризик захворіти на ХЛЛ становить приблизно 1:175 (0,57%). Цей ризик дещо вищий у чоловіків, ніж у жінок (1,3:1). ХЛЛ уражає в основному дорослих, середній вік на момент встановлення діагнозу становить близько 70 років. Хвороба рідко спостерігається в осіб до 40 років і надзвичайно рідко зустрічається у дітей [2]. ХЛЛ характеризується расовою схильністю: найчастіше зустрічається у представників європеоїдної раси, а найрідше – у монголоїдної (північно-східний вектор зменшення захворюваності), захворюваність у афроамериканців є проміжною.
Стандартизований показник захворюваності на ХЛЛ у розвинутих країнах коливається від 4 до 6 випадків на 100 тис. населення на рік, однак з віком захворюваність зростає і у старших вікових групах (80 років і старше) становить 30 на 100 тис. на рік.
В Україні на 1 січня 2019 р. було виявлено 1277 нових випадків захворювання на ХЛЛ, або 4,19 на 100 тис. населення, поширеність захворювання склала 10 092 випадки, або 33,15 на 100 тис. населення. На рисунку 1 наведено динаміку показників захворюваності та поширеності ХЛЛ в Україні за 2011-2019 рр. [3].
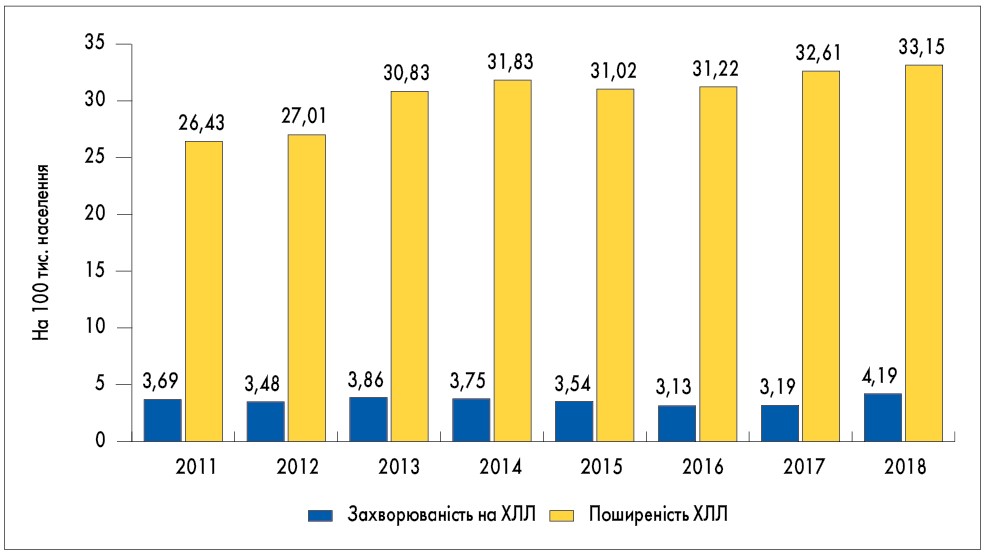 Рис. 1. Захворюваність і поширеність ХЛЛ серед дорослого населення України
Рис. 1. Захворюваність і поширеність ХЛЛ серед дорослого населення України
Клінічному перебігу ХЛЛ притаманні значні відмінності: від відсутності значимих клінічних проявів і життя пацієнтів впродовж десятиліть практично без лікування до швидкого та агресивного прогресування хвороби, що потребує ранньої та інтенсивної терапії. Основні фактори, котрі впливають на таку варіабельність захворювання, представлені на рисунку 2.
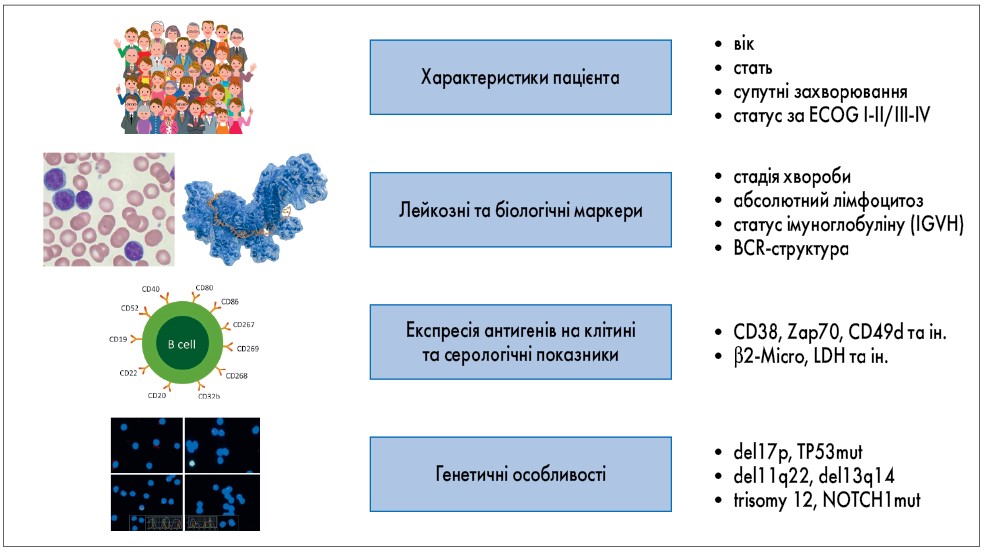 Рис. 2. Гетерогенність клінічних і біологічних характеристик ХЛЛ
Рис. 2. Гетерогенність клінічних і біологічних характеристик ХЛЛ
Як правило, пацієнти звертаються до гематолога після виявлення характерних змін у розгорнутому аналізі крові (в тому числі наявність анемії або тромбоцитопенії), за якими можна припустити наявність лейкозу, однак може бути і звернення з лімфаденопатією, спленомегалією або системними симптомами, такими як різка втомлюваність і слабкість, значна нічна пітливість, втрата маси тіла, тривале підвищення температури тіла.
ХЛЛ можна припустити на підставі підрахунку та морфологічного дослідження лімфоцитів периферичної крові, а саме – при виявлені характерних зрілих лімфоцитів у мазках крові й абсолютної кількості лімфоцитів >5,0×109/л. Для підтвердження діагнозу необхідно довести їх моноклональність, тобто походження від єдиної пухлинної клітини. Ця мета досягається імунофенотипуванням лейкоцитарних антигенів мононуклеарних клітин периферичної крові та/або кісткового мозку. У мінімальній панелі моноклональних антитіл для проведення імунофенотипування використовуються такі маркери: CD5, CD10, CD19, CD20, CD23, CD79b, CD38 κ (каппа), λ (лямбда).
Діагностичний алгоритм за останні кілька років істотно ускладнився і на сьогодні включає не тільки традиційні клініко-лабораторні й інструментальні методи обстеження для верифікації діагнозу та початку лікування, а також додаткові діагностичні опції.
До традиційного визначення стадії хвороби за Rai/Binet [4, 5], індексу коморбідності (Cumulative Illness Rating Scale – CIRS) [6], імуноцитофлуометричного/імуногістохімічного дослідження пухлини додаються дослідження сироваткових маркерів, молекулярно-генетичних/FISH (fluorescence in situ hybridization) особливостей лейкозу (для виявлення хромосомних аберацій t(11;14); t(11q; v); +12; del(11q); del(13q); del(17p), TP53), а також встановлення мутаційного статусу генів варіабельних ділянок важких ланцюгів імуноглобулінів (immunoglobulin heavy chain variable region – IGHV), що дає можливість розрахувати для пацієнта міжнародний прогностичний індекс CLL-IPI [7]. Загалом ці складні дослідження сприяють ретельному обґрунтуванню діагнозу, визначенню у пацієнта групи ризику та плануванню оптимальної лікувальної стратегії.
Перед початком хіміоімунотерапії з приводу лейкозу важливо дослідити згортальну систему крові пацієнта та провести тести на гепатити (маркери гепатитів В і С та кількісна полімеразна ланцюгова реакція гепатиту В). Необхідність цих обстежень випливає з тих міркувань, що проведення хіміоімунотерапії та індукований нею розпад тканин пухлини істотно впливають на систему гемостазу і можуть спровокувати значні, а інколи і фатальні тромбоемболії. Лікування препаратами моноклональних антитіл (імунотерапія) призводить до реактивації вірусу гепатиту В або до тяжкого перебігу вчасно не виявленого гепатиту В чи С. Тому при позитивних маркерах гепатитів обов’язкова консультація лікаря-інфекціоніста.
Дослідження кісткового мозку (аспірація або трепанобіопсія) не є обов’язковою процедурою для діагностування ХЛЛ, але для підтвердження повної відповіді після лікування таку маніпуляцію доцільно проводити. Також може виникнути необхідність виконання біопсії лімфатичних вузлів з подальшим морфологічним та імуногістохімічним дослідженням за припущення трансформації ХЛЛ в агресивну неходжкінську лімфому або лімфому Ходжкіна (ріхтерівської трансформації). Таку трансформацію можна запідозрити у разі різкої зміни перебігу лейкозу, відсутності відповіді на адекватне лікування, стрімкого розвитку нетипового рецидиву захворювання. Гематолог або онколог при оцінюванні перебігу ХЛЛ та появі особливостей в розвитку захворювання завжди має пам’ятати про можливу ріхтерівську трансформацію.
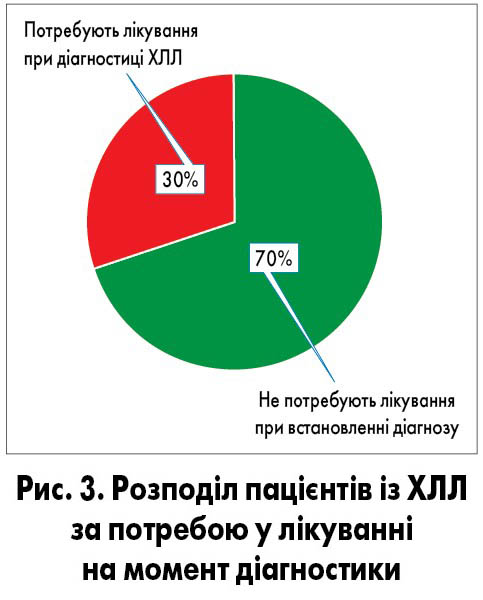 Переходячи до терапії ХЛЛ, слід ще раз наголосити, що це захворювання може проявлятися та перебігати по-різному, і тому при певному клінічному варіанті й особливостях ХЛЛ медикаментозне лікування може не знадобитися. Терапія може бути не потрібна безпосередньо після діагностики захворювання (рис. 3) для частини пацієнтів [8]. А деякі хворі (менше 20%) із встановленим діагнозом ХЛЛ взагалі ніколи не потребують лікування.
Переходячи до терапії ХЛЛ, слід ще раз наголосити, що це захворювання може проявлятися та перебігати по-різному, і тому при певному клінічному варіанті й особливостях ХЛЛ медикаментозне лікування може не знадобитися. Терапія може бути не потрібна безпосередньо після діагностики захворювання (рис. 3) для частини пацієнтів [8]. А деякі хворі (менше 20%) із встановленим діагнозом ХЛЛ взагалі ніколи не потребують лікування.
Дуже важливою проблемою ХЛЛ є вибір оптимального часу старту активного лікування. Хоча захворювання у своїй назві і містить термін «лейкоз», у 15-20% пацієнтів прогресування хвороби та потреби у лікуванні може не бути багато років. У кількох рандомізованих дослідженнях було продемонстровано, що початок лікування безпосередньо після встановлення діагнозу не збільшує загальну тривалість життя при ХЛЛ, тому терапію не слід починати за стабільного перебігу і мінімальних проявів ХЛЛ (0-I за Rai, стадія А за Binet) [9, 10].
І сьогодні дослідники дотримуються аналогічної думки про відтермінування терапії та дотримання тактики «дивись та чекай» (watch and wait) при початкових стадіях хвороби [11]. Як вважає Danielle M. Brander [12], передчасно проведена активна терапія може вибірково знищити менш агресивні лейкозні клітини, а клітини пухлини, які проліферують на їх місці після лікування, стають більш агресивними, тобто відбувається клональна еволюція.
Тому вкрай важливими для лікаря гематолога або онколога є оцінювання та дотримання розроблених міжнародною групою з вивчення ХЛЛ (iwCLL) критеріїв, наявність яких дозволяє розпочати активну медикаментозну терапію ХЛЛ [13]. Ці критерії (зі змінами та доповненнями) включають:
1. B-симптоми (симптоми лейкозної інтоксикації):
- втрата маси тіла >10% від попередньої за 6 місяців;
- значна втомлюваність (пацієнт може перебувати на амбулаторному лікуванні і бути здатним до самообслуговування, але не може виконувати будь-яку активну роботу);
- лихоманка більше 38 °C принаймні протягом 2 тижнів без ознак інфекційного процесу;
- виражене нічне потіння тривалістю більше 1 місяця без ознак інфекційного процесу.
2. Наявність прогресування недостатності кісткового мозку (інфільтрація моноклональними лімфоцитами), що проявляється низькими показниками крові (цитопенії), включно з анемією (рівень гемоглобіну <110 г/л) або тромбоцитопенією (кількість тромбоцитів <100×109/л).
3. Масивна або симптоматична спленомегалія (селезінка виступає >6 см нижче від ребрової дуги).
4. Масивні лімфатичні вузли або конгломерати вузлів (>10 см), прогресуюча або симптоматична лімфаденопатія (збільшені лімфатичні вузли порушують функцію органів).
5. Аутоімунна гемолітична анемія та/або імунна тромбоцитопенічна пурпура.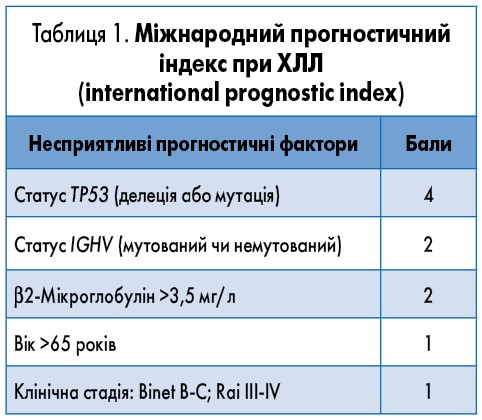
6. Прогресування лімфоцитозу: збільшення кількості лімфоцитів >50% за 2 місяці, час подвоєння лімфоцитозу <6 місяців. Якщо абсолютна кількість лімфоцитів становить <30 000×109/л, то час подвоєння лімфоцитів не слід використовувати як єдиний критерій початку лікування.
7. Гіперлейкоцитоз периферичної крові (лейкоцитів >200×109/л),
8. Масивна інфільтрація кісткового мозку патологічними лімфоцитами (>80%).
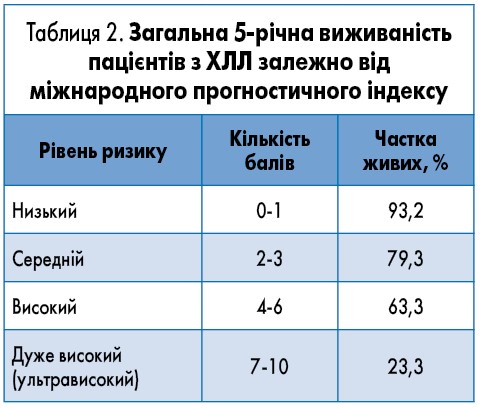
Перед початком терапії у пацієнтів з ХЛЛ важливо визначити групу ризику за CLL-IPI [14] (табл. 1).
За сумою набраних балів пацієнтів розподіляють на групи ризику, за якими можна оцінити загальну 5-річну виживаність пацієнтів (табл. 2) при умові надання їм сучасної ефективної медичної допомоги [14].
 Історично найдовше в терапії при ХЛЛ застосовується хлорамбуцил. Відповідь на лікування хлорамбуцилом спостерігалась у 70% хворих, однак повна ремісія – тільки у 2-10%. Хлорамбуцил і сьогодні використовують в окремих схемах лікування при ХЛЛ. На початку медикаментозної терапії ХЛЛ також застосовували циклофосфамід і кортикостероїди. Сьогодні використовується променева терапія на конгломерати лімфатичних вузлів та селезінку з метою зменшення пухлинної маси та компресійних симптомів. В окремих випадках, при вкрай великих розмірах селезінки та неефективності інших методів консервативної терапії, застосовуються і хірургічні методи лікування, а саме – спленектомія.
Історично найдовше в терапії при ХЛЛ застосовується хлорамбуцил. Відповідь на лікування хлорамбуцилом спостерігалась у 70% хворих, однак повна ремісія – тільки у 2-10%. Хлорамбуцил і сьогодні використовують в окремих схемах лікування при ХЛЛ. На початку медикаментозної терапії ХЛЛ також застосовували циклофосфамід і кортикостероїди. Сьогодні використовується променева терапія на конгломерати лімфатичних вузлів та селезінку з метою зменшення пухлинної маси та компресійних симптомів. В окремих випадках, при вкрай великих розмірах селезінки та неефективності інших методів консервативної терапії, застосовуються і хірургічні методи лікування, а саме – спленектомія.
З 1960-х років лікування ХЛЛ подолало шлях від використання простих лікарських засобів (хлорамбуцил, циклофосфамід) до складних інноваційних препаратів та методів лікування (рис. 4), включно з алогенною трансплантацією стовбурових гемопоетичних клітин та лікуванням Т-клітинами, так званою CARТ-терапією (Chimeric Antigen Receptor T-Cell).
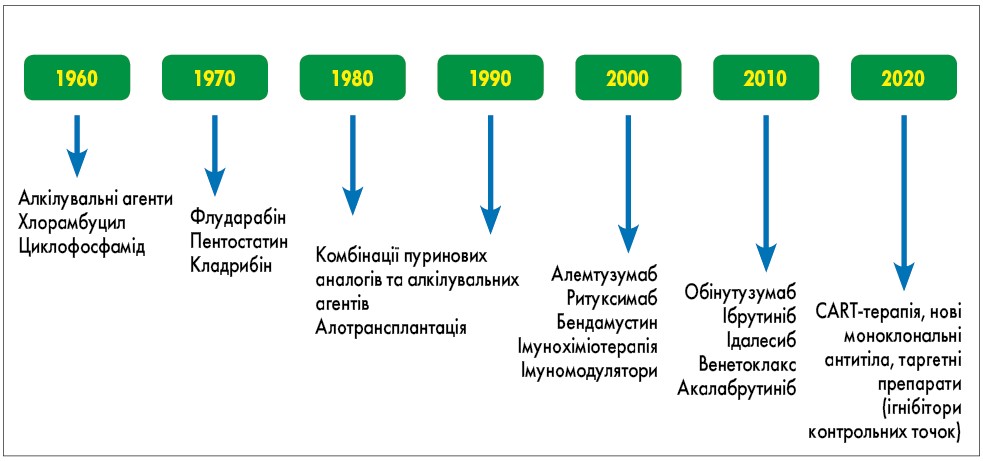 Рис. 4. Поява нових лікувальних опцій для застосування при ХЛЛ упродовж 60 років
Рис. 4. Поява нових лікувальних опцій для застосування при ХЛЛ упродовж 60 років
Метою цієї публікації не є ретельний розбір та обґрунтування усіх можливих сучасних опцій лікування ХЛЛ. Це досить складне завдання. Рішення щодо вибору способу лікування залежить від багатьох факторів, зокрема, віку пацієнта та його загального функціонального стану, супутніх захворювань, наявності del(17p) або мутації гена TP53, статусу IGHV (мутований чи немутований), попереднього лікування, доступності лікарських засобів, побажань пацієнта та інших.
В Україні наказом МОЗ від 12.05.2016 № 439 був затверджений Уніфікований клінічний протокол первинної, вторинної (спеціалізованої), третинної (високоспеціалізованої) медичної допомоги при хронічному лімфоцитарному лейкозі, відповідно до якого може надаватися медична допомога (рис. 5).
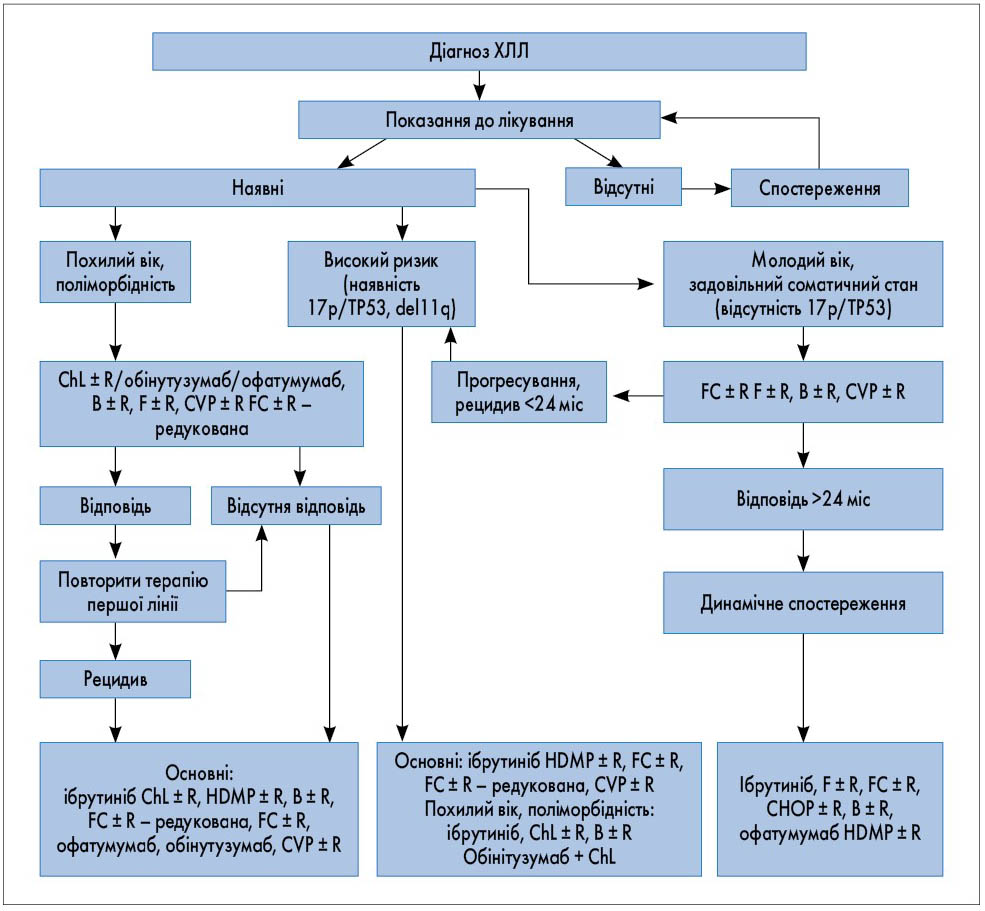 Рис. 5. Алгоритм надання допомоги при ХЛЛ відповідно до уніфікованого клінічного протоколу. CVР ± R може використовуватися при неможливості проведення іншого лікування
Рис. 5. Алгоритм надання допомоги при ХЛЛ відповідно до уніфікованого клінічного протоколу. CVР ± R може використовуватися при неможливості проведення іншого лікування
Однак спеціалізовані медичні заклади можуть надавати таку допомогу за своїми локальними протоколами, які ґрунтуються на адаптованих і затверджених міжнародних клінічних протоколах і настановах, наприклад Національної онкологічної мережі США (NCCN) [15] чи Європейського товариства медичної онкології (ESMO) [16]. Такі рекомендації періодично оновлюються відповідно до появи новітніх лікувальних опцій, і лікар, який слідкує за тенденціями в терапії ХЛЛ, завжди може знайти в цих документах відповіді щодо сучасного лікування пацієнтів.
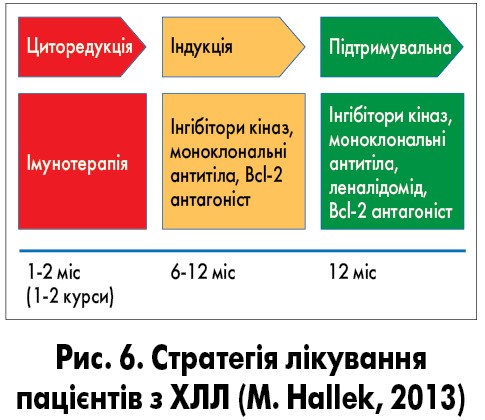 Ще у 2013 р. Michael Hallek [17] запропонував стратегію боротьби з ХЛЛ та досягнення тривалого контролю над хворобою (рис. 6). Ця стратегія послідовного потрійного Т (sequential triple-T: tailored, targeted, total eradication of MRD) передбачає лікувальний процес, спрямований практично на цілковитий контроль над хворобою, а саме на повну ерадикацію мінімальної залишкової хвороби (МЗХ). Автор зазначав, що, на його думку, у майбутньому лікарі після першого етапу (зменшення пухлинної маси) повинні переходити до другого етапу – використання цільових (таргетних) агентів, тобто нових нехіміотерапевтичних засобів з механізмом дії, спрямованим на патогенні сигнальні шляхи клітин ХЛЛ та їх мікрооточення. Результатом такого лікування стане повне знищення лейкозного клону, що можна буде оцінити за негативною МЗХ, визначеною майбутнім надчутливим методом.
Ще у 2013 р. Michael Hallek [17] запропонував стратегію боротьби з ХЛЛ та досягнення тривалого контролю над хворобою (рис. 6). Ця стратегія послідовного потрійного Т (sequential triple-T: tailored, targeted, total eradication of MRD) передбачає лікувальний процес, спрямований практично на цілковитий контроль над хворобою, а саме на повну ерадикацію мінімальної залишкової хвороби (МЗХ). Автор зазначав, що, на його думку, у майбутньому лікарі після першого етапу (зменшення пухлинної маси) повинні переходити до другого етапу – використання цільових (таргетних) агентів, тобто нових нехіміотерапевтичних засобів з механізмом дії, спрямованим на патогенні сигнальні шляхи клітин ХЛЛ та їх мікрооточення. Результатом такого лікування стане повне знищення лейкозного клону, що можна буде оцінити за негативною МЗХ, визначеною майбутнім надчутливим методом.
Третій етап стратегії потрійного Т має забезпечити збереження та підтримання дуже хорошої ремісії. Цього можна досягти, призначаючи пацієнтові монотерапію пероральним препаратом (типу інгібіторів кінази, Bcl‑2 або імуномодулятор) чи будь-яким іншим нехіміотерапевтичним засобом на тривалий період. Цей терапевтичний підхід контролюватиметься за допомогою оцінювання МЗХ, а лікування може бути припинено через декілька місяців (3-6-9‑12) після досягнення МЗХ‑негативної ремісії чи «перезапущене» у разі виявлення позитивної МЗХ. Цей етап може тривати довгий час для забезпечення максимального лікувального ефекту у найкоротші терміни і доти, доки гематологи й онкологи отримають історично унікальну можливість повністю контролювати поки що невиліковне захворювання – ХЛЛ.
Висновки:
- не всі пацієнти з ХЛЛ потребують лікування;
- деяким хворим ніколи не буде потрібне лікування;
- лікування ХЛЛ поступово буде зміщуватися в бік нехіміотерапевтичних засобів і методів;
- у майбутньому усі пацієнти з ХЛЛ, незалежно від віку та біології хвороби, будуть отримувати нехіміотерапевтичний лікарський засіб як терапію першої лінії;
- пошук нових лікарських засобів і методів лікування сприятиме повному та тривалому контролю над хворобою.
Раніше пацієнти з ХЛЛ жили для того, щоб постійно лікуватися, а сьогодні вони лікуються, щоб жити нормальним життям.
Література
- British Journal of Haematology. Volume 111, Issue 4, pages 1023-1034, 2 AUG 2008 DOI: 10.1111/j.1365-2141.2000.02215.
- Last Medical Review: May 10, 2018 Last Revised: January 8, 2020: https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/about/key-statistics.html.
- Новак В.Л., Масляк З.В., Горяінова Н.В. і співавт. Показники діяльності гематологічної служби науково-практичних підрозділів профільних ДУ НАМН України в 2018 році. 2018. – С. 52.
...
15. Hallek Michael Hallek. Signaling the end of chronic lymphocytic leukemia: new frontline treatment strategies. Blood. 2013 Nov 28; 122 (23): 3723-3734.
Тематичний номер «Онкологія. Гематологія. Хіміотерапія» № 1 (62), 2020 р.