


29 квітня, 2020
Врожденные и приобретенные нарушения гемостаза
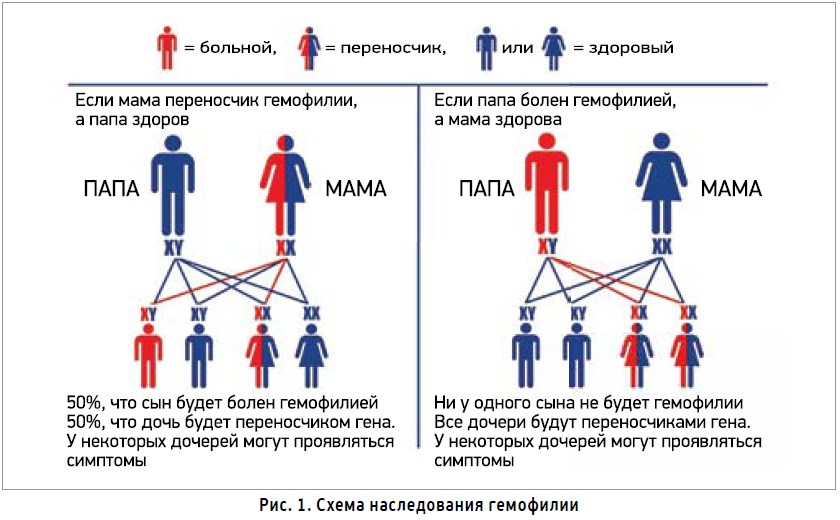
 Гемофилия – это наследственное заболевание, проявляющееся недостаточностью факторов свертываемости крови VIII (гемофилия типа А) или IX (типа В). Болезнь имеет Х-сцепленный тип наследования (рис. 1). Таким образом, подавляющее большинство больных гемофилией – мужского пола (у некоторых девочек могут появляться симптомы).
Гемофилия – это наследственное заболевание, проявляющееся недостаточностью факторов свертываемости крови VIII (гемофилия типа А) или IX (типа В). Болезнь имеет Х-сцепленный тип наследования (рис. 1). Таким образом, подавляющее большинство больных гемофилией – мужского пола (у некоторых девочек могут появляться симптомы).
Согласно эпидемиологическим данным Всемирной федерации гемофилии (The World Federation of Hemophilia – WFH), распространенность болезни в среднем составляет 10-14 случаев на 100 тыс. мужчин. Следует отметить, что, кроме наследственной передачи гена гемофилии, возможна спонтанная мутация, по некоторым источникам, обусловливающая до 30% случаев болезни. Около 70-80% всех случаев заболевания составляет гемофилия типа А, при которой наблюдается снижение активности фактора VIIІ (ФVIIІ), 6-13% – гемофилия типа В (вследствие снижения активности фактора IX – ФIX).
В зависимости от активности фактора свертывания крови выделяют 3 степени тяжести гемофилии. Нормальным считают уровень активности фактора свертывания крови от 50 до 150%. При тяжелой форме гемофилии (активность фактора свертывания крови составляет <1%), которая диагностируется у 60-70% больных, возникают спонтанные кровотечения, главным образом в суставы и мышечные ткани. Гемофилия средней тяжести (активность фактора свертывания крови – 1-5%) характеризуется сильными кровотечениями при травмах, хирургических вмешательствах, также возможны спонтанные кровотечения. При легкой форме болезни (активность фактора свертывания крови – 5-40%) сильные кровотечения возникают в случае серьезных травм или больших хирургических вмешательств.
По локализации кровоизлияний у пациентов с гемофилией доминируют гемартрозы (70-90%), гематомы, экхимозы (10-20%), реже встречаютсяо – другие локализации (5-10%) и кровотечения в центральной нервной системе (менее 2%).
При ведении пациентов с гемофилией перед врачом стоит задача остановить кровотечение и предотвратить его рецидивы. До настоящего времени главным методом лечения гемофилии остается заместительная терапия соответствующим дефицитным фактором свертывания.
Согласно рекомендациям WFH, выделяют следующие варианты лечения:
- экстренная терапия – лечение острых состояний;
- профилактическое лечение – первичная и вторичная профилактика;
- домашнее лечение;
- домашнее лечение «по требованию»;
- ортопедическая реконструктивная терапия;
- хирургическое лечение;
- физиотерапия и лечебная физкультура.
В мире первичная профилактика является основным направлением терапии у пациентов до 17 лет. У взрослых пациентов преобладает терапия «по требованию». Приверженность к указанному виду терапии частично обусловлена проблемами, связанными с венозным доступом. Проведение профилактического лечения дает возможность минимизировать инвалидизацию пациентов с гемофилией или вовсе избежать ее. Начинается профилактическое лечение после первого гемартроза в раннем детском возрасте. При тяжелом течении заболевания рекомендуется продолжать лечение как можно дольше вне зависимости от возраста. Внедрение профилактического лечения позволяет пациентам с гемофилией жить полноценной жизнью и избежать инвалидизации.
До 1960-х годов для лечения больных гемофилией использовали цельную кровь или замороженную плазму. Позднее появились криопреципитат, а затем – лиофилизированный концентрат ФVIII, что существенно упростило лечение пациентов с гемофилией А. После внедрения диагностических тестов на вирусы гепатита С и иммунодефицита человека начали использовать вирусинактивированный концентрат ФVIII, а клонирование гена ФVIII инициировало появление первого рекомбинантного ФVIII. С накоплением новых знаний разработаны новые препараты, в частности ингибиторные комплексы, рекомбинантный фактор VIIa (ФVIIa), факторы с пролонгированным периодом полувыведения, что позволяет уменьшить частоту их внутривенного введения. Сегодня активно изучаются возможности использования моноклональных антител и генных препаратов в лечении больных гемофилией (Franchini M. et al., 2013; Berntorp E. et al., 2012).
Для заместительной терапии используются плазменные и рекомбинантные факторы свертывания. Несмотря на тройную вирусинактивацию плазменных факторов свертывания, риск передачи гемотрансмиссивных инфекций (в частности, прионов) остается. Как плазменные, так и рекомбинантные факторы свертывания вызывают возникновение ингибиторов. Обе группы имеют такие преимущества, как возможность использования в домашних условиях, точный подбор дозы. Однако в случае применения рекомбинантных факторов свертывания полностью отсутствует риск передачи гемотрансмиссивных инфекций и есть возможность получения неограниченного количества препарата. Так, например, при использовании плазменных факторов свертывания в качестве профилактического лечения гемофилии для одного пациента с массой тела 75 кг на 1 год терапии в среднем необходимо произвести 1237 заборов донорской крови по 700 мл для получения 866 л плазмы (Burnouf T., 2007). В этом аспекте возможность производства рекомбинантных факторов свертывания имеет большое значение.
Согласно данным WFH, выделяют 4 группы ожидаемых последствий для пациентов в зависимости от количества международных единиц (МЕ) ФVIII на душу населения. При <1 МЕ ФVIII ожидается сокращение продолжительности жизни пациентов, при 1 МЕ – сохранение побочных эффектов, приводящих к социальной дезадаптации, при 2-4 МЕ возможно проведение надлежащего лечения рецидивов, плановых оперативных вмешательств и вторичной профилактики поражений суставов, при 5-7 МЕ – первичной профилактики и индукции иммунной толерантности (Evatt B. L., 2002). В 2018 году в Украине зафиксировано 1,13 МЕ ФVIII на душу населения, что является критически низким уровнем.
Самым тяжелым осложнением, связанным с лечением гемофилии, является образование ингибитора к ФVIII/ФIX. При этом состоянии образуются антитела, которые прикрепляются к ФVIII или ФIX и нейтрализуют либо подавляют его способность к остановке кровотечения. Клинически это проявляется отсутствием ответа на стандартное лечение концентратами факторов свертывания, что делает заместительную терапию неэффективной.
Около 99% случаев образования ингибиторов приходится на гемофилию А, остальные – на гемофилию В. (Долгое время считалось, что при гемофилии В ингибиторы вообще не возникают.) В соответствии с эпидемиологическими данными WFH ингибиторную форму гемофилии имеют около 20-30% пациентов с тяжелым течением болезни и до 3-5% – со среднетяжелым. Ингибиторы образуются в среднем после 50-150 введений ФVIII. Следует отметить, что риск возникновения ингибитора выше у пациентов с отягощенным аллергологическим анамнезом. Наиболее часто ингибитор развивается в первые 50 дней введения фактора и после интенсивной терапии при хирургическом вмешательстве, чаще – у детей, поэтому перед проведением хирургического лечения пациента с гемофилией важно определить уровень ингибитора, например с помощью лабораторного теста смешивания плазмы.
В связи с неэффективностью заместительной терапии факторами свертывания тактика ведения пациентов с ингибиторной формой заболевания кардинально отличается. У пациентов с низким титром ингибитора кровотечение может быть купировано введением концентрата фактора в дозах, превосходящих стандартные в 3 раза (индукция иммунной толерантности – ITI-терапия) (уровень доказательности IIIB). В отсутствие эффекта необходимо применение препаратов шунтирующего действия. Остановка кровотечений у пациентов с высоким титром ингибитора должна проводиться только препаратами шунтирующего действия: антиингибиторным коагулянтным комплексом или активированным рекомбинантным ФVII (эптаког альфа).
Кроме наследственной гемофилии, в клинической практике встречается приобретенная гемофилия – спонтанно развивающееся аутоиммунное заболевание, причиной которого является формирование блокирующих антител к ФVIII свертывания крови (Knoebl P. et al., 2012). Распространенность заболевания составляет 1,5 случая на 1 млн населения в год (рис. 2).
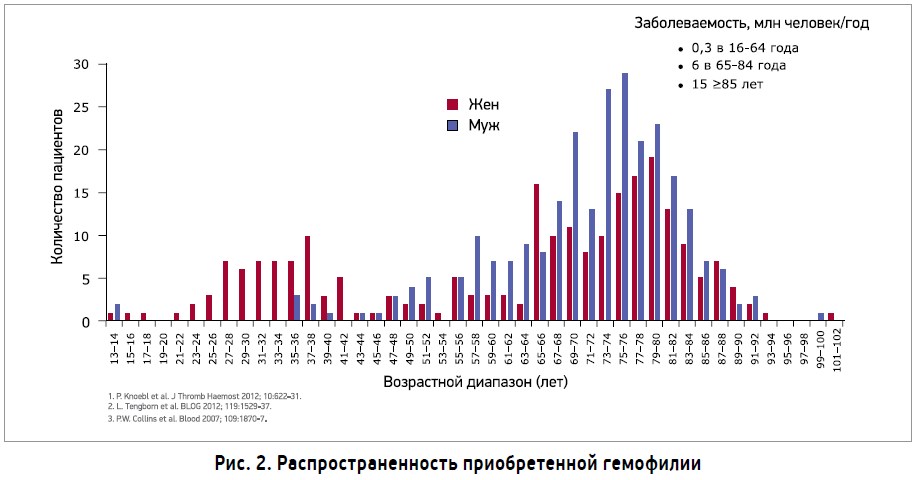
Кровотечения имеют иной характер, чем при наследственной гемофилии, обычно возникают спонтанно. При приобретенной гемофилии кровотечения возникают чаще в подкожной жировой клетчатке, коже, мышцах, желудочно-кишечном тракте, реже развиваются генитальные, ретроперитонеальные кровотечения, кровоизлияния в суставы и др. (Collins P. W. et al., 2007; Delgado J.l. et al., 2003). Приобретенная гемофилия в 50% случаев ассоциирована с рядом состояний: аутоиммунными болезнями, злокачественными новообразованиями, заболеваниями кожи, гиперчувствительностью к лекарственным препаратам, беременностью. Приблизительно у 18% женщин приобретенная гемофилия ассоциирована с беременностью и развивается в послеродовой период на 3-150-е сутки в виде массивного кровотечения. Пик распространенности приобретенной гемофилии приходится на возраст 69-80 лет, но среди женщин патология также часто встречается в детородном возрасте (Collins P. et al., 2010; Borg J. Y. et al., 2013; Scharf R. E. et al., 2011; Zeitler H. et al., 2010; Knoebl P. et al., 2012; Green D. et al., 1981; Morrison A. E. et al., 1993).
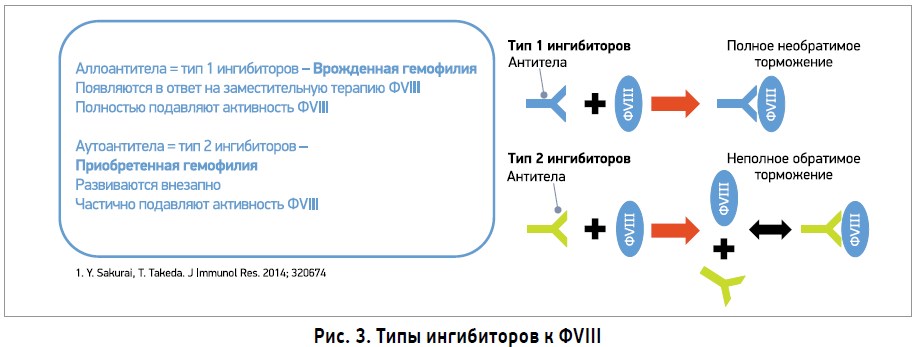
Выделяют 2 типа ингибиторов к ФVIII (рис. 3). Аллоантитела (1 тип) наблюдаются у пациентов с врожденной гемофилией. Эти антитела инактивируют ФVIII с линейной скоростью, связанной с их концентрацией, и могут полностью угнетать ФVIII при высоких концентрациях. Aутоантитела (2 тип) выявляют у пациентов с приобретенной гемофилией. Эти антитела имеют нелинейный ингибирующий профиль, развиваются внезапно и частично угнетают ФVIII (Sakurai Y. et al., 2014).
Терапевтическая тактика при приобретенной гемофилии преследует две цели: остановить кровотечение (в случае острого кровотечения, требующего вмешательства) и устранить ингибиторы (сразу после верификации диагноза приобретенной гемофилии). Для остановки кровотечения используют рекомбинантный ФVIIа, антиингибиторный комплекс, препараты ФVIII (при низком уровне ингибитора).
Последняя опция в данном перечне имеет невысокую эффективность, и во всем мире врачи отдают предпочтение рекомбинантному ФVIIа, который является более надежным методом достижения гемостаза. Эрадикация ингибиторов осуществляется за счет иммуносупрессивной терапии (преднизолон, ритуксимаб, азатиоприн). Не менее важным также является лечение основного заболевания.
Сегодня на фармацевтическом рынке активированный рекомбинантный фактор коагуляции VII эптаког альфа представлен препаратом НовоСевен® (компания Novo Nordisk). Лекарственное средство используется для лечения кровотечений и их профилактики при хирургических вмешательствах либо при других инвазивных процедурах у пациентов с врожденной гемофилией с уровнем ингибиторов к ФVIII или ФIX >5 BU либо выраженной реакцией на введение ФVIII или ФIX в анамнезе, приобретенной гемофилией, врожденным дефицитом фактора VII, тромбастенией Гланцмана с антителами к GP IIb-IIIa и/или HLA и резистентностью к переливанию тромбоцитарной массы в прошлом либо в настоящее время.
Препарат НовоСевен® применяется в начальной дозе 90 мкг (4,5 КМО) на 1 кг массы тела каждые 2-3 ч до достижения гемостаза. При необходимости продолжения лечения после достижения эффективного гемостаза введение повторяют через 4, 6, 8 или 12 ч (в зависимости от клинической ситуации). Дальнейшее лечение может быть целесообразным для предотвращения повторного кровотечения. Данная схема лечения представлена в соответствии с инструкцией к применению препарата НовоСевен® в Украине.
Доказательная медицина
На основе Регистра мониторинга аутоантител у пациентов с приобретенной гемофилией было проведено проспективное контролируемое исследование, в рамках которого проанализированы данные о распространенности и течении болезни, ассоциации гемофилии с другими патологическими состояниями, а также результаты гемостатической и эрадикационной терапии у 82 больных. Возраст более ⅔ пациентов составлял >70 лет, около 50% случаев гемофилии были связаны с другими заболеваниями, 46% пациентов получали гемостатическую терапию рекомбинантным ФVIIа, у 81% из них кровотечение было купировано или уменьшено (Borg J. Y. et al., 2013).
Также был проведен анализ Регистра Общества исследований гемофилии и тромбоза и других источников, который включил данные 139 пациентов и 182 эпизода кровотечения. При назначении рекомбинантного VIIа в качестве первой линии терапии лечение было эффективным в 95% случаев, во второй линии – в 80% (Sumner M. J. et al., 2007).
Таким образом, при подозрении на патологию гемостаза необходимо провести исследования, включающие определение уровня факторов свертывания в крови и уровня ингибитора. При планировании хирургического вмешательства у пациентов с нарушением гемостаза целесообразно создание мультидисциплинарной группы с включением гематолога.
Приобретенная гемофилия встречается гораздо чаще, чем диагностируется, и информированность врачей поможет сохранить настороженность в отношении этого заболевания.
Медична газета «Здоров’я України 21 сторіччя» № 7 (476), квітень 2020 р.