


10 липня, 2016
Метформин: новые горизонты применения
Бигуанид метформин – наиболее широко используемый при сахарном диабете (СД) 2 типа препарат, его принимают около 150 млн пациентов во всем мире. Метформин был получен из Galega officinalis (галега лекарственная, козья рута, французская сирень), традиционного лекарственного растения, широко используемого в средневековой Европе для облегчения симптомов диабета. В конце XIX в. было установлено, что G. officinalis содержит большое количество гуанидина – соединения, у которого впоследствии были обнаружены гипогликемические свойства. Однако клиническому применению гуанидина препятствовала его выраженная токсичность, и исследователи переключили свой фокус на более безопасные аналоги. Бигуаниды, состоящие из двух N-связанных гуанидинов, были синтезированы в 1920-х гг., однако их терапевтический потенциал остался без надлежащего внимания вследствие появления в том же десятилетии инсулинотерапии. И только в 1957 г., после публикации успешного исследования французского врача Жана Стерна, метформин начал использоваться для лечения диабета. В отличие от ранее применявшихся противодиабетических средств преимуществом бигуандидов была способность снижать уровни глюкозы в крови без риска развития гипогликемии. Первоначально популярными были более сильные бигуаниды фенформин и буформин, однако после большого количества сообщений об ассоциированном лактатацидозе эти препараты в 1970-х гг. были отозваны с рынка в большинстве стран. В итоге благодаря хорошему профилю безопасности метформин стал препаратом первой линии терапии СД 2 типа и сегодня входит в перечень жизненно важных лекарств Всемирной организации здравоохранения. В настоящее время очевидно, что терапевтический потенциал метформина выходит далеко за рамки лечения диабета. Появляется все больше данных, демонстрирующих роль препарата в терапии множества заболеваний, включая рак и кардиоваскулярную патологию. Кроме того, имеются доказательства того, что метформин замедляет процесс старения и модулирует микробиоту, которая, как известно, имеет огромное значение для поддержания здоровья человека. В настоящем обзоре обсуждаются предложенные молекулярные механизмы, объясняющие эти разнообразные эффекты.
Метформин и СД 2 типа
Согласно данным статистики, в мире в настоящее время проживает примерно 382 млн пациентов с СД. Из-за урбанизации и связанного с ней роста распространенности ожирения и малоподвижного образа жизни в 2035 г. эта цифра, по прогнозам, достигнет 592 млн. СД 2 типа, на который приходится 85-95% случаев диабета, характеризуется гипергликемией, возникающей вследствие инсулинорезистентности или нарушенной секреции инсулина. Это заболевание с комплексной этиологией, включающей взаимодействие между множеством генетических и внешних факторов. Сильными предикторами СД 2 типа являются отягощенный семейный анамнез, повышенный индекс массы тела, высокое артериальное давление, низкая физическая активность, нездоровое питание и старший возраст. В долгосрочной перспективе СД 2 типа может приводить к тяжелым и опасным для жизни осложнениям, таким как кардиоваскулярные заболевания, нейропатия, ретинопатия и нефропатия. Развитие этих осложнений можно предотвратить или значительно отсрочить посредством эффективного контроля уровня глюкозы в крови, для достижения которого необходима модификация образа жизни и, во многих случаях, назначение пероральных антигипергликемических препаратов, таких как метформин.
Несмотря на то что метформин применяется в лечении диабета с 1957 г., в настоящее время открываются все новые и новые механизмы его действия. Раньше антигипергликемический эффект препарата связывали с повышением чувствительности гепатоцитов к инсулину и повышенным захватом глюкозы периферическими тканями. Сегодня установлено, что метформин действует преимущественно путем супрессии глюконеогенеза в печени. У пациентов с диабетом метформин может снижать глюконеогенез на 36%. Тем не менее молекулярные механизмы, ответственные за снижение продукции глюкозы, остаются предметом обсуждений. Предложенные механизмы действия метформина при СД 2 типа представлены на рисунке 1.
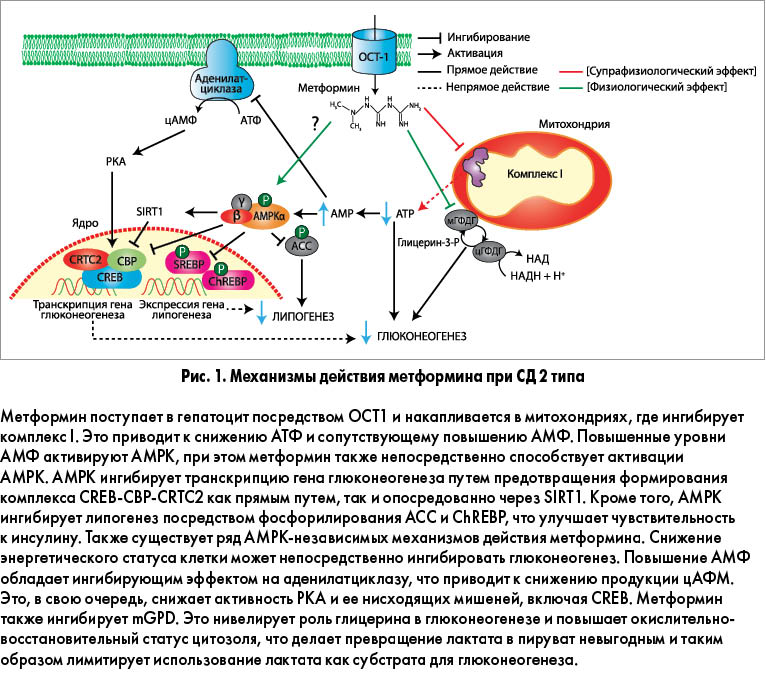
Молекулярные мишени метформина
Благодаря необычной гидрофильной природе метформин не может пассивно проникать через клеточные мембраны; его поступление в гепатоциты осуществляют органические катионные транспортеры (OCT). Установлено, что терапевтическая эффективность метформина обеспечивается транспортером ОСТ1, при этом генетический полиморфизм ОСТ1 у человека может обусловливать вариабельный ответ на препарат. После захвата гепатоцитом метформин накапливается в митохондриальном матриксе. Считается, что ключевой мишенью метформина является комплекс I дыхательной цепи митохондрий. Этот вывод был сделан по результатам двух независимых работ, показавших, что метформин селективно ингибирует окисление субстратов комплекса I, но не субстратов комплексов II или IV. Эти данные, изначально полученные на культурах изолированных гепатоцитов крыс, впоследствии были подтверждены на многочисленных клеточных моделях первичных гепатоцитов человека.
Механизм ингибирования метформином комплекса I точно не установлен; предполагается, что повышенный уровень рН митохондриального матрикса превращает метформин в депротонированную форму с высокой аффинностью к ионам меди. Эти медные комплексы могут взаимодействовать с чувствительными окислительно-восстановительными реакциями дыхательной цепи, что согласуется с более ранними наблюдениями о зависимости клеточных эффектов метформина от способности препарата связывать медь. Альтернативный механизм ингибирования комплекса I недавно был описан Bridges и соавт. Исследователи продемонстрировали, что метформин неконкурентно ингибирует восстановление убихинона, вероятно, посредством связывания с интерфейсом гидрофильных и мембранных доменов, а также связывания этого фермента в неактивную конформацию с открытой петлей.
Следствием ингибирования комплекса I метформинов является снижение продукции аденозинтрифосфата (АТФ) с сопутствующим повышением уровней аденозинмонофосфата (АМФ) и аденозиндифосфата (АДФ). Этот сдвиг энергетического метаболизма клетки детектируется главным энергетическим сенсором клеток – AMP-активированной протеинкиназой (AMPK).
AMPK-зависимые механизмы
AMPK – эффективный регулятор энергетического гомеостаза клеток, который активируется путем связывания молекул АДФ или АМФ с сайтом его регуляторной γ-субъединицы. Это позволяет клетке отвечать на сниженный энергетический статус переходом с АТФ-потребляющего анаболического состояния в АТФ-продуцирующее катаболическое состояние.
Ключевая роль AMPK в механизме действия метформина была показана в 2001 г. после публикации известного исследования Zhou и соавт. Ученые установили, что метформин стимулирует активацию AMPK в первичных гепатоцитах крыс, и использовали ингибитор AMPK соединение С, чтобы продемонстрировать необходимость AMPK для ингибирования продукции глюкозы под действием метформина. Впоследствии эти данные были подтверждены Saw и соавт., обнаружившими, что утрата печеночной киназы В1 (LKB1), которая является восходящим активатором AMPK, отвечающим за фосфорилирование ее каталитической α-субъединицы, исключает глюкозоснижающие эффекты метформина у мышей, находящихся на богатой жирами диете.
Было высказано предположение, что сигнальный путь LKB1/AMPK, активирующийся метформином, изменяет программу глюконеогенеза клетки путем ингибирования опосредуемого циклическим аденозинмонофосфатом (цАМФ) ответа коактиватора транскрипции 2 (CRTC2), регулируемого элементсвязывающим белком (CREB), – ключевого регулятора экспрессии гена глюконеогенеза). CRTC2 располагается в ядре клетки, где он соединяется с CREB и обеспечивает повышающую регуляцию коактиватора 1α рецептора γ, активируемого пролифератором пероксисом (PGC-1α), и его нисходящих целевых генов фосфоенолпируваткарбоксикиназы и глюкозо-6-фосфата. Деактивация CRTC2 также может достигаться путем AMPK-опосредуемой индукции никотинамидфосфорибозилтрансферазы и сопутствующего повышения печеночного сиртуина 1 (SIRT1). Деацетилирование CRTC2 под действием SIRT1 l делает его уязвимым к убиквитинированию, опосредуемому фотоморфогенезом 1, и дегредации, следствием чего является ингибирование экспрессии гена глюконеогенеза. Также было установлено, что AMPK запускает диссоциацию транскрипционного комплекса CREB-CREB-связующий белок (СВР)-CRTC2 путем фосфорилирования CBP в позиции Ser436.
Значимость статуса AMPK как основного медиатора действия метформина была поставлена под сомнение после публикации результатов работы Foretz и соавт. В этом исследовании было установлено, что метформин ингибирует продукцию глюкозы у трансгенных мышей, у которых отсутствуют печеночные каталитические субъединицы AMPK или LKB1. Авторы высказали предположение, что несоответствие между полученными ими результатами и предыдущими наблюдениями Saw и соавт. может быть обусловлено тем, что в последнем исследовании прямой эффект метформина на продукцию глюкозы в печени не оценивался, а изучалось влияние повторного назначения метформина на уровни глюкозы крови натощак. Следовательно, наблюдения Saw и соавт. в действительности могут быть отражением непрямого эффекта на ось AMPK-LKB1 продукции глюкозы в печени, вероятно, опосредуемого супрессией липогенеза под действием AMPK. Хорошо изученной мишенью AMPK является ацетил-КоА-карбоксилаза (АСС) – скоростьлимитирующий фермент, необходимый для образования малонил-КоА, который, в свою очередь, является предшественником липогенеза и ингибитором β-окисления. У мышей ингибирование АСС под действием AMPK регулирует индуцируемое метформином улучшение чувствительности к инсулину. Кроме того, AMPK может вызывать понижающую регуляцию экспрессии ряда липогенных генов. Таким образом, AMPK может отвечать за долгосрочный терапевтический эффект метформина, достигаемый посредством благоприятной модификации не только углеводного, но и липидного метаболизма, что способствует повышению чувствительности к инсулину.
Большинство исследователей полагают, что метформин активирует AMPK путем повышения соотношения АДФ/АМФ посредством ингибирования митохондриального дыхания, однако существуют и другие гипотезы. Например, метаболические изменения, вызываемые метформином в изолированных клетках скелетных мышц, не согласуются с нарушением продукции энергии в митохондриях; скорее, они отражают прямое ингибирование фермента АМФ-деаминазы, что, в свою очередь, приводит к повышению уровней АМФ и активации AMPK. Также существует возможность того, что AMPK сама по себе является прямой мишенью метформина. В частности, установлено, что метформин в низких концентрациях может повышать образование АМФ-αβγ-трехмерного гетеротрехмерного комплекса в гепатоцитах и in vitro, повышая фосфорилирование каталитической α-субъединицы, хотя потенциальные сайты связывания метформина пока обнаружить не удалось. С другой стороны, в ряде исследований было продемонстрировано, что метформин не является прямым аллостерическим активатором. Таким образом, ингибирование дыхательной цепи с сопутствующим повышением уровней АМФ в настоящее время рассматривается как наиболее вероятный механизм, объясняющий активацию AMPK под действием метформина.
AMPK-независимые механизмы
Учитывая неоднозначность статуса AMPK как ключевого медиатора действия метформина, был предложен ряд AMPK-независимых механизмов. Один из них состоит в том, что ассоциированные изменения клеточного энергетического метаболизма могут непосредственно модулировать продукцию глюкозы. Глюконеогенез – энергетически затратный процесс, в котором для синтеза 1 молекулы глюкозы требуется 6 молекул АТФ. Поскольку терапия метформином приводит к снижению уровней АТФ, гепатоцитам приходится соответствующим образом снижать продукцию глюкозы. Так, Foretz и соавт. установили сильную корреляцию между снижением содержания АТФ и ингибированием продукции глюкозы в первичных мышиных гепатоцитах, инкубируемых с метформином, что свидетельствует о тесной взаимосвязи энергетического статуса гепатоцитов и продукции глюкозы в печени. Кроме того, изменения энергетического статуса клеток, вызываемые метформином, могут подавлять глюконеогенез путем аллостерического ингибирования эссенциальных ферментов. Например, АМФ действует синергически с фруктозо-2,6-бисфосфатом, ингибируя ключевой фермент глюконеогенеза фруктозо-1,6-бифосфатазу.
Наличие у метформина молекулярных механизмов действия, не зависящих от транскрипционных изменений, подтверждается в исследованиях по изучению экспрессии генов. Форсированная экспрессия PGC-1α (главного коактиватора генов глюконеогенеза) не препятствует проявлению метформин-индуцированного снижения продукции глюкозы в гепатоцитах. В ряде исследований также было установлено отсутствие корреляции между экспрессией генов глюконеогенеза и продукцией глюкозы в печени как на экспериментальных моделях у мышей, так и у пациентов с СД 2 типа. В целом результаты этих работ свидетельствуют о том, что ингибирование экспрессии генов глюконеогенеза не является основной детерминантой клинических эффектов метформина; последние также достигаются благодаря изменениям энергетического метаболизма клеток и ассоциированному снижению глюконеогенеза.
Другой AMPK-независимый механизм действия метформина, включающий антагонизм в отношении эффектов глюкагона, был недавно предложен Miller и соавт. В серии экспериментов in vitro и in vivo на первичных гепатоцитах мышей было продемонстрировано, что метформин и еще один бигуанид фенмормин блокируют глюкагон-индуцированную активацию аденилатциклазы, что приводит к снижению синтеза цАМФ. Это, в свою очередь, снижает активность протеинкиназы А (PKA), предотвращая фосфорилирование критических субстратов глюконеогенеза, таких как 6-фосфофрукто-2-киназа / фруктозо-2,6-бифосфатаза 1, CREB-1 и рецептор инозитолтрифосфата. По мнению исследователей, повышение внутриклеточных уровней АМФ, индуцируемое метформином, отвечает за ингибирование аденилатциклазы, вероятно, путем прямого связывания аденинового остатка АМФ с ингибиторным Р-сайтом.
Тем не менее следует отметить, что если антагонизм к глюкагону был бы основным механизмом действия метформина, применение этого препарата ассоциировалось бы с частыми эпизодами гипогликемии, как это наблюдается у трансгенных мышей, не имеющих рецепторов глюкагона. Однако одним из основных преимуществ метформина является очень низкая частота гипогликемии, поэтому его потенциальный антагонизм к глюкагону у человека, по-видимому, не полный или уменьшается компенсаторными механизмами.
Исследования, на которых основаны вышеуказанные модели AMPK-независимых эффектов метформина, подвергались критике за то, что в них использовались концентрации препарата, значительно превышающие максимальные терапевтические у пациентов с диабетом. Метформин назначается перорально, максимальная рекомендованная доза составляет 2,5 г/сут. Было продемонстрировано, что после приема однократный дозы 1,5 г пиковая концентрация в плазме у человека достигает примерно 4 мкг/мл, или 18 мкмоль. В то же время данные, полученные в экспериментах на животных, показали, что концентрация метформина в портальной вене печени значительно превышает таковую в системной плазме.
В исследованиях Foretz и соавт. и Miller и соавт. для демонстрации AMPK-независимых эффектов метформина действительно использовались высокие концентрации препарата, однако это не означает, что указанные эффекты не будут проявляться на клинически значимом уровне при применении более низких концентраций. В экспериментах in vivo на грызунах метформин часто назначается в дозах 250-350 мг/кг. Эти значения были получены с использованием стандартного метода конвертации доз между видами, основанного на нормализации площади поверхности тела. Согласно данной формуле стандартная терапевтическая доза 20 мг/кг у взрослого человека (масса тела 60 кг) эквивалентна дозе примерно 250 мг/кг у мыши. Кроме того, установлено, что у мышей с диабетом для проявления терапевтического эффекта метформин приходится назначать в относительно высоких дозах. Также продемонстрировано, что метформин накапливается в очень высоких концентрациях во многих тканях. У мышей доза 50 мг/кг создает концентрации более 250 мкмоль в печени и еще более высокие концентрации в тонком кишечнике. Таким образом, в исследованиях in vitro на первичных гепатоцитах совершенно оправдано использование концентраций метформина, превышающих таковые в плазме или портальной вене. Кроме того, известно, что биологические эффекты метформина являются время- и концентрационно-зависимыми, что, по-видимому, отражает свойство препарата накапливаться в митохондриальном матриксе. Это означает, что эффекты, наблюдаемые при высоких концентрациях, также могут проявиться и при более низких концентрациях после более продолжительного воздействия метформина.
Исследования, в которых для изучения AMPK-независимых механизмов действия метформина использовались нокаутированные по печеночной AMPK животные, следует интерпретировать с учетом недавно полученных доказательств важной роли тонкокишечной AMPK в реализации эффектов метформина. Главным местом действия метформина считается печень, однако активация AMPK в тонком кишечнике вносит существенный вклад в реализацию быстрого глюкозоснижающего эффекта препарата. В экспериментальном исследовании на инсулинорезистентных крысах было продемонстрировано участие оси «пищеварительный тракт – головной мозг – печень»: под действием метформина рецептор AMPK-глюкагоноподобного пептида-1 в двенадцатиперстной кишке передает сигналы в центральную нервную систему, которая, в свою очередь, дает «команду» печени снизить продукцию глюкозы. Результаты этого исследования также показали, что активация AMPK в органах, как правило, при более высоких концентрациях метформина (в милимолярном диапазоне), может отдаленно регулировать метаболизм тканей, непосредственно отвечающих за продукцию глюкозы.
Недавно был обнаружен новый механизм действия метформина, не зависящий ни от активации AMKP, ни от изменений энергетического статуса клетки. Madiraju и соавт. установили, что метформин ингибирует фермент глицерофосфатного шаттла митохондриальную глицерофосфатдегидрогеназу (mGPDG). Это предотвращает прямое использование глицерина как субстрата для глюконеогенеза, а также приводит к повышению окислительно-восстановительного статуса цитозоля, что делает невыгодным превращение лактата в пируват и таким образом ограничивает роль лактата в глюконеогенезе. Это исследование подчеркивает значимость метформина как неспецифического препарата, способного влиять на множество молекулярных мишеней с итоговым подавлением глюконеогенеза.
Метформин и кардиоваскулярные заболевания
К кардиоваскулярным заболеваниям (КВЗ) относятся болезни сердца и кровотока. По данным Британского фонда сердца (BHF), в Великобритании КВЗ являются причиной более четверти всех летальных исходов и обусловливают экономические потери в сумме 19 млрд фунтов стерлингов в год. Одним из самых сильных факторов риска развития КВЗ является СД 2 типа. Имеются доказательства, что метформин может защищать от КВЗ пациентов с диабетом. Это особенно важно с учетом того, что КВЗ являются ведущей причиной смерти больных СД. В масштабном исследовании UKPDS лечение метформином снизило риск инфаркта миокарда (ИМ) на 39% по сравнению с традиционной терапией за 10-летний период. Последующие исследования подтвердили благоприятную роль метформина в защите против кардиоваскулярных осложнений СД. Безусловно, эти положительные эффекты метформина частично могут быть обусловлены улучшением метаболизма глюкозы и липидов, однако существует и ряд других механизмов, объясняющих непосредственное влияние препарата на кардиоваскулярную систему.
Результаты многочисленных исследований свидетельствуют о том, что метформин обладает антиатеросклеротическими эффектами за счет улучшения целостности эндотелия и предотвращения формирования атеросклеротических бляшек. В частности, было установлено, что активация AMPK под действием метформина уменьшает повреждение эндотелиоцитов, вызываемое окислительным стрессом на фоне гипергликемии. Данный процесс опосредуется ингибированием сигнального пути протеинкиназы С-NAD(P)Н-оксидазы. В результате снижение продукции активных форм кислорода (АФК) в цитозоле останавливает инициацию петли положительной обратной связи между образованием АФК в митохондриях и расщеплением последних, что, в свою очередь, предотвращает эндотелиальный апоптоз путем снижения мембранного потенциала митохондрий.
В условиях инсулинорезистентности метформин также проявляет антитромботические свойства. Например, метформин противодействует стимулирующему эффекту гиперинсулинемии на продукцию ингибитора активатора плазминогена 1 (PAI-1) – отрицательного регулятора фибринолиза, принимающего участие в образовании кровяного сгустка. Предполагается, что этот эффект опосредуется не улучшением чувствительности к инсулину, ассоциированным с метформином, а непосредственным ингибированием экспрессии гена PAI-1. Кроме того, у пациентов с инсулинозависимым диабетом лечение метформином значительно уменьшает агрегацию тромбоцитов. Это наблюдение может объясняться результатами экспериментального исследования, в котором было обнаружено, что метформин AMKP-зависимо усиливает фосфорилирование Ser1179 эндотелиальной синтазы оксида азота (NO), что улучшает биодоступность NO для эндотелия крупных сосудов. Как известно, NO играет ключевую роль в поддержании сосудистого гомеостаза, выполняя множество функций, включая ингибирование агрегации тромбоцитов, и таким образом вносит существенный вклад в защиту от развития ишемической болезни сердца (ИБС).
Помимо эндотелийпротекторных эффектов метформин воздействует на функцию кардиомиоцитов, что в экспериментальных исследованиях ассоциировалось с уменьшением диабетической кардиомиопатии. Последняя является ведущей причиной сердечной недостаточности и характеризуется сосудистой дисфункцией, развивающейся независимо от ИБС и артериальной гипертензии. Высказывалось предположение, что метформин может корригировать нарушения релаксации кардиомиоцитов путем тирозинкиназозависимой модуляции кальция. Впоследствии было продемонстрировано, что уменьшение диабетической кардиомиопатии при лечении метформином ассоциируется с AMPK-опосредованной повышающей регуляцией аутофагии. Кардиальная аутофагия является важным механизмом гомеостаза, подавляющим диабетическую кардиомиопатию. AMPK, активируемая метформином, защищает кардиомиоциты путем разрушения комплекса беклин-1-Blc-2 (известного также как комплекс В-клеточной лимфомы 2) и переключения процессов апоптоза на аутофагию. В экспериментальных исследованиях на мышах с диабетом восстановление аутофагии улучшало структуру и функцию сердца.
Имеются доказательства, что метформин проявляет кардиопротекторные эффекты в отношении повреждения вследствие ишемии-реперфузии после ИМ. Назначение метформина на протяжении первых 15 мин после реперфузии ограничивало размеры инфаркта в изолированных сердцах крыс с диабетом и без диабета. Этот эффект опосредуется активацией сигнального пути протеинкиназы В (Akt) / фосфатидилинозитол-4,5-бифосфат-3-киназы (PI3K), что предотвращает открытие митохондриальной поры переходной проницаемости (mPTP) – ключевого триггера клеточной смерти во время реперфузии. Уменьшение размеров инфаркта при лечении метформином также объясняется AMPK-опосредованным повышением активности эндотелиальной синтазы NO. Интересно, что новый низкомолекулярный активатор AMPK (A-76922) действует синергически с метформином, что проявляется в усилении кардиопротекторных эффектов активации AMPK.
Метформин может уменьшать повреждение, обусловленное ишемией-реперфузией, не только в остром периоде, но и в долгосрочной перспективе. Так, в экспериментальном исследовании назначение метформина на протяжении 4 нед уменьшало размеры ИМ независимо от наличия диабета. При этом кардиопротекторные эффекты, наблюдавшиеся у животных с диабетом, ассоциировались с улучшением структуры митохондрий, вероятно, за счет активации AMPK и повышения экспрессии PGC-1α. Длительное лечение метформином после ИМ также может оказывать положительный эффект. Было установлено, что 12-недельная терапия метформином после ИМ уменьшает ремоделирование сердца и таким образом может отсрочивать развитие сердечной недостаточности.
Разнообразные положительные эффекты метформина на кардиоваскулярную систему, в том числе при диабете, в настоящее время изучаются в рандомизированных контролированных исследованиях.
Метформин и рак
Во всем мире злокачественные новообразования являются ведущей причиной смерти. Проведенный недавно в Великобритании анализ показал, что более чем у 50% людей в возрасте до 65 лет в тот или иной период жизни диагностируется рак. Высокая социально-экономическая значимость онкопатологии заставляет расходовать огромные средства на исследования новых методов лечения. Несмотря на это, только 5% противоопухолевых препаратов, достигающих I фазы клинических исследований, в итоге получают одобрение, и даже наиболее современные таргетные препараты лишь незначительно улучшают выживаемость.
Интерес к метформину в области онкологии возник в 2005 г. после публикации эпидемиологического исследования, в котором была установлена связь между лечением этим препаратом и сниженным риском рака у пациентов с диабетом. В последующем в многочисленных наблюдательных исследованиях подтвердилась протекторная роль метформина в отношении различных злокачественных опухолей, включая гепатоцеллюлярную карциному, колоректальный рак, рак желудка и пищевода, в диабетической популяции. Способность метформина изменять метаболизм опухолевых клеток и взаимодействовать с разнообразными метаболическими путями свидетельствует о том, что этот препарат может быть эффективным в предотвращении развития и замедлении прогрессирования различных типов рака. Предполагаемые механизмы противоопухолевых эффектов метформина представлены на рисунке 2.
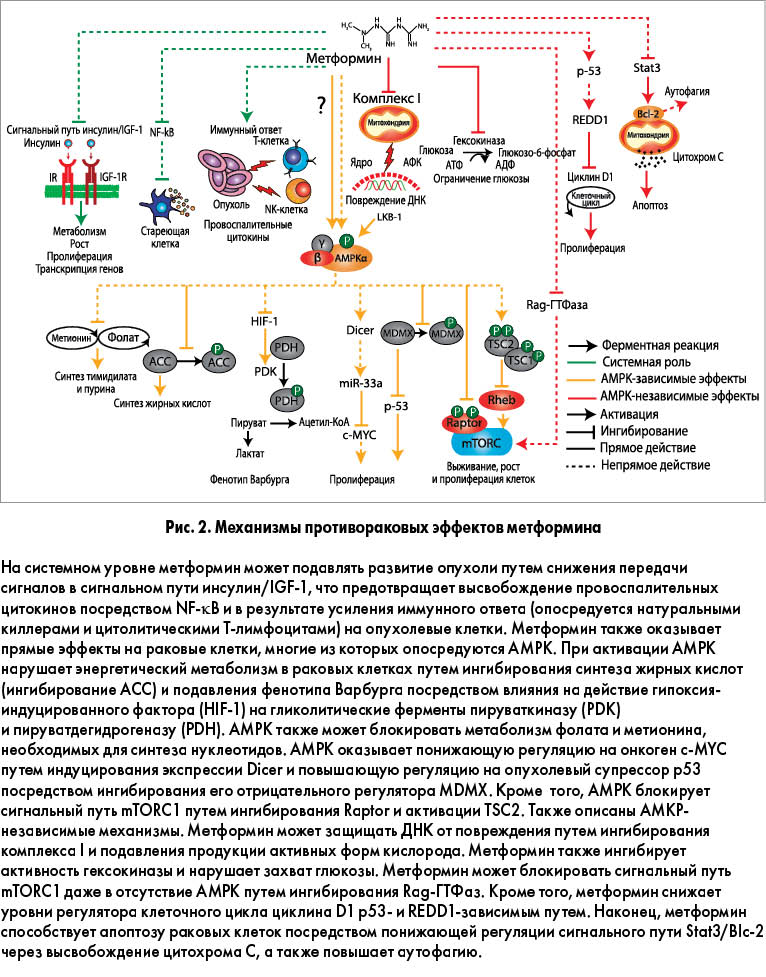
Возможно, защиту от рака обеспечивают системные эффекты метформина. В экспериментальных и эпидемиологических исследованиях было продемонстрировано, что инсулин и инсулиноподобный фактор роста 1 (IGF-1) могут способствовать развитию злокачественных опухолей за счет стимулирования пролиферации эпителиальных клеток. Метформин может предотвращать непластическую активность путем снижения гиперинсулинемии и уровней этих сигнальных молекул. Кроме того, метформин может модифицировать воспалительные процессы, принимающие участие в прогрессировании рака. Например, установлено, что метформин блокирует активность ядерного фактора транскрипции каппа В (NF-κB), уменьшая секрецию провоспалительных цитокинов стареющими клетками. Этот механизм также объясняет антиэйджинговые свойства препарата. Недавно было продемонстрировано, что метформин усиливает иммунный ответ на раковые клетки, в частности защищает опухоль-инфильтрирующие лимфоциты CD8+ от апоптоза и функционального истощения. Многообещающей находкой стало также то, что метформин способен повышать эффективность экспериментальной противоопухолевой вакцины путем улучшения выживаемости Т-клеток памяти.
AMPK-зависимые механизмы
Противоопухолевые эффекты метформина в значительной степени опосредуются активацией сигнального пути LKB1/AMPK. LKB1 является опухолевым супрессором, и мутация гена LKB1 ассоциируется с синдромом Пейта-Йегерса – врожденным заболеванием с предрасположенностью к злокачественным новообразованиям. Было продемонстрировано, что протекторный эффект метформина ослабляется при фармакологическом ингибировании AMPK и в экспериментальных моделях у AMPK-нокаутированных животных. Активация AMPK приводит к подавлению передачи сигналов в пути mTOR (мишени рапамицина млекопитающих) – нутриентчувствительного регулятора синтеза белка, роста и пролиферации клеток. Этот эффект достигается путем прямого фосфорилирования Ser1345 и Thr1227 опухолевого супрессора TSC2 (белка туберозного склероза 2), который формирует mTOR-комплекс 1 (mTORC1) с TSC1. Кроме того, AMPK может предотвращать активацию mTORC1 посредством фосфорилирования его связующего партнера Raptor.
Активация AMPK сопровождается подавлением эффекта Варбурга – приобретаемого раковыми клетками метаболического фенотипа, который характеризуется предпочтением аэробного гликолиза над окислительным фосфорилированием. Эта трансформация облегчается гипоксия-индуцибельным фактором 1α – транскрипционным фактором, активирующим экспрессию гликолитического гена в ответ на передачу сигналов mTORC1. Классической метаболической характеристикой рака, связанной с эффектом Варбурга, является повышенный синтез жирных кислот de novo и повышенные уровни ключевого липогенного фермента – синтетазы жирных кислот (FAS), описанные при различных типах рака у человека. Установлено, что активация AMPK снижает экспрессию FAS в клетках рака предстательной железы и уменьшает их выживаемость. В экспериментальном исследовании на модели карциномы толстой кишки было продемонстрировано, что метформин снижает экспрессию FAS и противодействует стимулирующему влиянию высококалорийной пищи на рост опухоли. Эти данные свидетельствуют о том, что метформин может проявлять свои противоопухолевые эффекты путем изменения метаболизма глюкозы и липидов, что в конечном итоге ограничивает доступ раковых клеток к субстратам, необходимым для роста.
Недавно в нескольких работах было установлено, что метформин может действовать как антифолатный препарат, нарушая фолатный цикл подобно аналогичному эффекту антифолатных химиотерапевтических агентов. Фолатный, или одноуглеродный, цикл является ключевым регулятором клеточного метаболизма и интегратором нутриентного статуса. Он ассимилирует глюкозу и аминокислоты, которые посредством химических реакций трансформируются для выполнения различных биологических функцией. Последние включают клеточный биосинтез, регуляцию кислотно-основного состояния, регуляцию эпигенетики путем метилирования нуклеиновых кислот и белков, сохранение генома посредством регуляции пула нуклеотидов. Например, фолатный цикл регулирует de novo синтез нуклеотидов, необходимых для починки и репликации ДНК, а также для продукции универсального донатора метильной группы S-аденозилметионина, принимающего участие в различных реакциях метилирования, в частности метилирования ДНК. Химиотерапевтические препараты, такие как метотрексат, аминопретин и азасерин, блокируют фолатный метаболизм и нарушают de novo синтез тимидилатных и пуриновых нуклеотидов. Установлено, что метформин нарушает метаболизм фолата и метионина у Escherichia coli, а также в клетках рака грудной железы с одновременным снижением метаболитов глутатиона и триптофана.
Помимо вышеописанных изменений метаболизма метформин может регулировать клеточный цикл путем взаимодействия с классическими онкогенами и опухолевыми супрессорами. Например, было продемонстрировано, что метформин вызывает AMPK-зависимую понижающую регуляцию c-MYC в клеточных линиях рака грудной железы. Этот эффект опосредуется повышенной экспрессией miR-33a (микроРНК, которая связывается с регионом 3’UTR с-MYC), что, в свою очередь, вызывается повышающей регуляцией фермента РНКазы III Dicer в ответ на лечение метформином. Модуляция Dicer под действием метформина имеет особую значимость с учетом того, что низкие уровни этого фермента ассоциируются с неблагоприятным прогнозом при раке яичников, грудной железы и легких.
AMPK-независимые механизмы
Описан также ряд AMPK-независимых механизмов, объясняющих противоопухолевые свойства метформина. В частности, метформин может защищать ДНК от повреждений и мутаций, подавляя продукцию АФК комплексом I. Кроме того, может активировать mTORC1 в отсутствие AMPK, что достигается ингибированием Rag-ГТФаз, отвечающих за индуцирование транслокации mTORC1 в клеточные компартменты, содержащие активатор mTORC1 – Rheb (гомолог Ras, концентрирующийся в головном мозге). Антипролиферативные эффекты метформина при раке предстательной железы связаны с AMPK-независимым ингибированием критического регулятора клеточного цикла циклина D1. Предполагается, что данный процесс опосредуется р53-зависимой повышающей регуляцией REDD1 (белка, регулируемого при эмбриогенезе и повреждении ДНК). Исследование, проведенное на клетках плоскоклеточного рака пищевода, показало, что метформин способствует их апоптозу и аутофагии, эффективно подавляя рост опухоли. Этот эффект был обусловлен инактивацией сигнального пути Stat3 (сигнальный трансдуктор и активатор транскрипции 3) / Bcl2. На другой клеточной модели были продемонстрировано, что метформин нарушает захват глюкозы клетками рака легкого путем аллостерического ингибирования гексокиназы-II. Дефицит этого энергетического субстрата вызывает деполяризацию митохондрий с последующим апоптозом. Аналогичный эффект также наблюдался в отношении клеток рака грудной железы.
Комбинированная противоопухолевая терапия с включением метформина
Получены доказательства, что метформин улучшает ответ раковых клеток на лучевую терапию, а также повышает чувствительность различных типов раковых клеток к широко применяющимся химиотерапевтическим средствам, включая цисплатин, паклитаксел, карбоплатин и доксорубицин. Однако следует отметить, что метформин снижал цитотоксический эффект цисплатина на клеточных линиях глиомы, нейробластомы, фибросаркомы и лейкоза посредством AMPK-независимой регуляции сигнального пути выживания Akt. Метформин может быть особенно эффективным при использовании в комбинации с низкомолекулярными ингибиторами киназы. На клеточных линиях меланомы было продемонстрировано, что эффективность ингибиторов протеинкиназы BRAF (гомолога В1 вирусного онкогена мышиной саркомы v Raf) ограничивается компенсаторным окислительным фосфорилированием. Соответственно, ингибиторы окислительного фосфорилирования повышают эффективность этих препаратов. В отличие от многих ингибиторов окислительного фосфорилирования, проявляющих выраженную токсичность, метформин имеет благоприятный профиль безопасности и, следовательно, высокий терапевтический потенциал.
Метформин и старение
Биологическая основа процесса старения остается главной загадкой для науки, однако за последние годы на пути к ее решению был достигнут значительный прогресс. Генетические, диетические и фармакологические вмешательства могут улучшать здоровье и продлевать жизнь ряда лабораторных организмов, таких как нематода Caenorhabditis elegans, плодовая муха Drosophila melanogaster и домовая мышь Mus musculus. Эти вмешательства ослабляют процесс старения и увеличивают продолжительность жизни, защищая от возрастных патологических изменений. Например, контролированное снижение потребления пищи без нутритивной недостаточности продлевает жизнь многим животным. Метформин может имитировать этот эффект и таким образом обеспечивать схожие преимущества у человека.
Бигуаниды и выживаемость C. elegans
В многочисленных исследованиях было установлено, что бигуаниды могут поддерживать и продлевать жизнь C. elegans. В одной из первых таких работ назначение буформина в дозе 0,1 мг/мл на протяжении жизни (включая личиночную стадию) увеличивало среднюю и максимальную продолжительность жизни взрослого червя на 23 и 26% соответственно. Назначение метформина на протяжении жизни в дозе 50 ммоль увеличивало среднюю продолжительность жизни на 40%. Интересно, что более низкие и более высокие дозы метформина (10 и 100 ммоль соответственно) не имели подобного эффекта. В двух недавних исследованиях было продемонстрировано, что эффекты метформина в дозе 50 ммоль не зависят от его назначения на ранних стадиях развития и увеличение выживаемости также достигается при использовании препарата в более поздние периоды жизни. Аналогичным образом фенформин в концентрациях 1,5; 3 и 4,5 ммоль продлевал жизнь взрослых червей на 5, 21 и 26% соответственно. Так как же бигуаниды увеличивают продолжительность жизни C. elegans?
Метформин обладает комплексным механизмом действия, вызывая широкий спектр физиологических эффектов. Установлено, что способность препарата продлевать жизнь в значительной степени связана с его влиянием на метаболизм фолата и метионина бактерий-симбионтов. Кроме того, метформин действует как прямой метаболический стрессор (вероятно, путем ингибирования комплекса I дыхательной цепи), активируя ответ на окислительный стресс и детоксикацию с вовлечением ряда метаболических медиаторов, включая AMPK и SKN-1. Это подтверждается тем, что назначение антиоксидантов, таких как N-ацетилцистеин, препятствующих продукции АФК и мутациям AMPK/SKN-1, нарушает развитие адекватного детоксицирующего ответа, индуцируемого метформином путем влияния на бактериальный метаболизм.
Метформин может не только продлевать жизнь, но и увеличивать продолжительность здоровой жизни C. elegans. Так, меторфмин снижает накопление пигмента молекулярного повреждения липофусцина, увеличивает подвижность на поздних периодах жизни, снижает накопление жировой ткани, ослабляет возрастные морфологические изменения и повышает выживаемость особей, подвергающихся длительной аноксии.
Бигуаниды и выживаемость насекомых
У насекомых эффекты бигуанидов на выживаемость имеют свои особенности. Назначение метформина в дозах 1; 2,5 и 5 ммоль не влияло на выживаемость мужских и женских особей D. melanogaster, при этом более высокие дозы (100 ммоль у самцов; 25, 50 и 100 ммоль у самок) снижали этот параметр, несмотря на выраженную активацию AMPK и снижение жировых запасов. Этот результат противоречит данным других работ, где было продемонстрировано, что повышенные уровни экспрессии и активации AMPK увеличивают выживаемость C. elegans и D. melanogaster.
Хорошо описанной особенностью предиабета у человека является инфекционно-индуцируемая гипергликемия. На модели ожирения у D. melanogaster назначение метформина в дозе 10 ммоль снижало неблагоприятные последствия инфекции Rhizopus – гриба, вызывающего мукормикоз у пациентов с диабетом. Метформин уменьшал набор веса, нормализовал уровни глюкозы, повышенные вследствие инфекции, и повышал выживаемость. При назначении в дозе 5 ммоль метформин замедлял возрастные изменения кишечных стволовых клеток D. melanogaster, что проявлялось в снижении молекулярных маркеров повреждения ДНК gH2AX foci и 8-oxodG. В одном из последних исследований, выполненных на домовых сверчках Acheta domesticus, метформин увеличивал среднюю и максимальную продолжительность жизни у 23,2% мужских и 43,7% женских особей.
Бигуаниды и выживаемость грызунов
Результаты многочисленных исследований, проведенных на грызунах, указывают на эволюционно сохранившуюся жизнепродлевающую роль бигуанидов. Назначение метформина мышам линий HER-2/neu, SHR, 129/Sv, C57BL/6 и B6C3F1 в широком спектре дозировок увеличивало продолжительность жизни на 4-38%. При этом установлено, что данный эффект препарата зависит от генотипа, пола, используемого бигуанида и его дозы, а также от длительности лечения. В целом, чем позже было начато лечение, тем менее выраженным было продление жизни (схожая зависимость наблюдалась и в экспериментах с C. elegans). Например, назначение мышам SHR метформина в 15-месячном возрасте не увеличивало продолжительность жизни по сравнению с назначением в 3-месячном возрасте (0 vs 38% соответственно). У мышей C57BL/6 доза метформина 0,1% от суммарного рациона продлевала жизнь на 5,83%, тогда как доза 1% – сокращала ее на 14,4%; уменьшение продолжительности жизни было обусловлено нефротоксичностью. У мышей 129/Sv назначение метформина продлевало жизнь на 5% у самок, но не у самцов. Интересно, что влияние бигуанидов на продолжительность жизни крыс менее выраженно по сравнению с мышами.
Замедление старения и продление жизни лабораторных животных при назначении метформина, по-видимому, обусловлено широким спектром эффектов препарата, включая изменения веса и температуры, нормализацию сывороточных уровней глюкозы, инсулина, триглицеридов и холестерина, регуляцию эстрогенной функции и антинеопластические эффекты. Недавно в исследовании Martin-Montalvo и соавт. было установлено, что метформин, имитируя эффекты ограничения энергетической ценности рациона, играет важную роль в продлении жизни и сохранении здоровья. Метформин улучшает фитнес, повышает чувствительность тканей к инсулину, снижает уровни липопротеинов низкой плотности и холестерина без снижения калорийности пищи. Интересно, что повышение активности AMPK при назначении метформина происходит без сопутствующих изменений в дыхательной цепи. Кроме того, метформин активирует антиоксидантные и противовоспалительные ответы, что уменьшает окислительное повреждение тканей и хроническое воспаление.
В целом накопленные экспериментальные данные сформировали базис для клинического применения метформина как антиэйджингового препарата. Продолжение исследований в этой области, очевидно, имеет огромные перспективы.
Метформин и увеличение продолжительности жизни человека
На сегодня проспективные исследования, в которых оценивалось бы влияние метформина на выживаемость здоровых людей, не проводились. В 10-летнем рандомизированном клиническом исследовании метформина у пациентов с избыточной массой тела или ожирением, страдающих СД 2 типа (UKPDS-O), были продемонстрированы долгосрочные благоприятные эффекты препарата на здоровье и выживаемость, при этом наблюдалось снижение кардиальной и общей смертности у пациентов, получавших метформин. Важно отметить, что метаболические преимущества от лечения метформином длительно сохраняются даже после его отмены. Тем не менее молекулярные механизмы, посредством которых метформин проявляет отдаленные эффекты на выживаемость, не связанные с гликемическим контролем, остаются неизученными. Интересно, что в исследовании UKPDS-S, продолжавшемся 6,6 года, комбинированная терапия меторфмином и производным сульфонилмочевины (ПСМ) по сравнению с монотерапией ПСМ в разнородной группе пациентов с СД 2 типа (как с нормальной массой тела, так и с избыточным весом и ожирением) проявляла потенциально негативные эффекты. Учитывая эти данные и результаты экспериментальных работ, для лучшего понимания долгосрочных благоприятных эффектов метформина на продолжительность жизни человека целесообразно проведение новых исследований, нацеленных на здоровую и относительно молодую популяцию.
Метформин и микробиота
Практически все живые организмы не живут в изоляции, а формируют тесные взаимоотношения с другими видами, прежде всего микроорганизмами. Кишечник является домом для большинства микробов, населяющих тело человека, и эта широкая популяция бактерий, которую называют «забытым органом», принимает участие во многих метаболических процессах с важными последствиями для физиологии и модуляции метаболических фенотипов человека. На сегодня доказана связь между изменениями кишечной микробиоты (вызванными диетой, антибиотикотерапией и т. п.) и развитием таких заболеваний, как ожирение, СД 2 типа, метаболический синдром и онкопатология, а также ускоренным старением, т. е. микробиота может служить биомаркером здоровья макроорганизма. Следовательно, препараты, широко применяющиеся в лечении метаболических заболеваний, ассоциированных с дисфункцией микробиоты, могут влиять на бактериальные популяции и таким образом улучшать здоровье человека (рис. 3). Вопрос о том, в какой степени изменения микробиоты объясняют благоприятные эффекты метформина при метаболических заболеваниях, остается открытым. Предполагается, что этот препарат может модулировать состав и функции бактерий, населяющих гастроинтестинальный тракт.
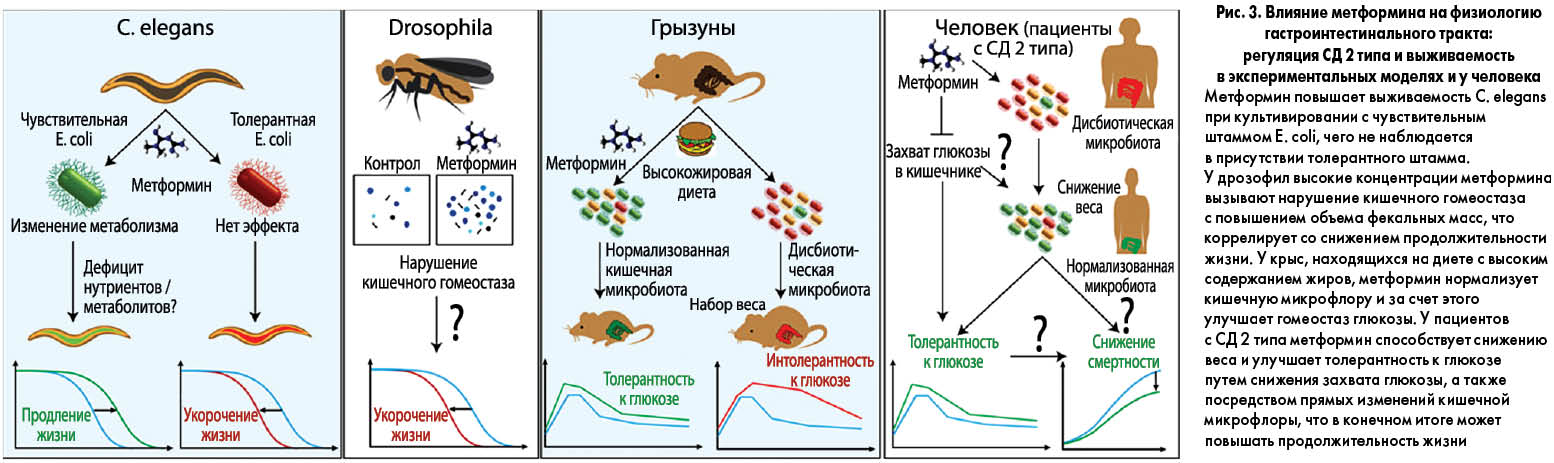
Метформин изменяет метаболизм и регулирует физиологию C. elegans
Нематода C. elegans сформировала выгодные взаимоотношения с E. coli, отражающие, хотя и в упрощенном виде, подобные ассоциации «здоровых» кишечных бактерий у человека. Было установлено, что у С. elegans метформин изменяет бактериальный метаболизм фолата и метионина, имитируя действие ограничения энергетической ценности пищи. При этом метформин замедляет процесс старения только в присутствии E. coli. Интересно, что у человека метформин также может вызывать дефицит фолата и витамина В12 с сопутствующим повышением уровней гомоцистеина.
Метформин влияет на кишечный гомеостаз у насекомых
Пищеварительный тракт D. melanogaster является одним из наиболее изученных органов этого насекомого. Как и у людей, задержка и абсорбция жидкости, перистальтика, кишечный транзит, частота дефекации и характер стула у дрозофил подвергаются сложной гомеостатической регуляции. В предыдущих исследованиях было установлено, что микробиом D. melanogaster модулирует развитие и метаболический гомеостаз организма-хозяина посредством влияния на сигнальный путь инсулина, тем не менее точная роль бактерий пока не определена.
Влияние метформина на функцию пищеварительного тракта у грызунов
Исследования на мышах показали, что метформин накапливается в стенке кишки в очень высоких концентрациях, и это может объяснять эффекты препарата на организм хозяина. В целом результаты исследований на грызунах указывают на то, что кишечник является главным органом, регулирующим действие метформина. Установлено, что метформин не только замедляет абсорбцию глюкозы в пищеварительном тракте, но и способствует ее утилизации путем неокислительного анаэробного метаболизма. Лактат как продукт анаэробного метаболизма по портальной вене может поступать в печень и там утилизироваться как субстрат для глюконеогенеза. Следовательно, кишечник играет важную роль в глюкозоснижающих эффектах метформина и одновременно служит защитным механизмом, предотвращающим гипогликемию.
Большинство микробов, сосуществующих с организмом-хозяином, находятся в пищеварительном тракте и поэтому подвергаются селективному воздействию высоких концентраций метформина. СД 2 типа и ожирение характеризуются структурными и функциональными изменениями популяций кишечных микробов, воспалительным состоянием и нарушением барьерной функции кишечника. Схожесть эффектов микробиоты и метформина на ожирение и регуляцию глюкозы крови указывает на существование общих механизмов, при этом противодиабетические эффекты метформина достигаются как прямым путем, так и опосредованно благодаря влиянию на кишечную микрофлору.
В двух недавно завершившихся исследованиях было установлено, что у мышей, находящихся на диете с высоким содержанием жиров, метформин улучшает метаболические маркеры и приближает состав фекальной микробиоты к таковому у животных, получающих нормальное питание. С помощью метагеномного анализа было продемонстрировано, что метформин вызывает глубокий сдвиг специфических подгрупп бактерий, в частности увеличивает содержание микроорганизмов рода Akkermansia. Кроме того, метформин повышает количество бокаловидных клеток, продуцирующих муцин, который является источником питания для Akkermansia municiphila. Известно, что назначение этой бактерии в качестве пробиотического препарата ассоциируется с улучшением метаболического профиля, снижением метаболической эндотоксемии и воспаления жировой ткани, улучшением гликемического контроля и снижением инсулинорезистентности; все эти эффекты также свойственны метформину.
У мышей, получающих обогащенную жирами пищу, метформин также увеличивает популяцию лактобацилл. Lactobacillus является одной из многих бактерий, которые могут утилизировать глюкозу с образованием лактата. Примечательно, что улучшение гомеостаза глюкозы при назначении метформина существенно ослабляется при сопутствующем применении антибиотиков широкого спектра действия. Эти данные свидетельствуют о том, что кишечная микробиота является терапевтической мишенью для метформина. По-видимому, повышенные уровни лактата и других субстратов для глюконеогенеза (пирувата, аланина) при лечении метформином не только защищают макроорганизм от гипогликемии, но и модифицируют структуру микробиоты, предоставляя новые энергетические субстраты определенным бактериям.
В исследовании на крысах было установлено, что комбинированная терапия пребиотическими пищевыми волокнами, такими как олигофруктоза, и метформином улучшает метаболический профиль у животных, находящихся на диете с высоким содержанием жиров. При этом олигофруктоза и метформин проявляли синергическое и аддитивное благоприятное действие на массу тела, печеночные триглицериды, секрецию глюкозозависимого инсулинотропного полипептида и лептина, а также на активацию AMPK, при этом значительно увеличивалось содержание бифидогенных бактерий, таких как Bifidobacterium spp.
Влияние метформина на кишечник и кишечную микрофлору человека
Терапевтические дозы большинства противодиабетических препаратов, использующихся у пациентов с СД 2 типа, находятся в диапазоне микрограмм и миллиграмм. В то же время эффективные дозы метформина могут достигать 2,5 г/сут. Тонкий кишечник является основным местом всасывания и аккумуляции метформина у животных и человека. Время абсорбции Т1/2 составляет 0,9-2,6 ч и зависит от принимаемой пищи, которая может отсрочивать этот процесс примерно на 35 мин. Метформин является стабильным и не связывается с белками, биодоступность составляет 50-60%. После перорального приема метформин достигает максимальной концентрации в периферической плазме через 2 ч, относительно быстро элиниминируется (период полувыведения – 1,7-4,5 ч) и экскретируется в неизмененном виде с калом и мочой. Как и у грызунов, метформин создает высокие концентрации в стенке тонкого кишечника человека, в 30-300 раз превышающие концентрации в плазме и других тканях. Концентрация метформина в просвете кишки после приема дозы 850 мг у пациентов с диабетом составляет около 20 ммоль. В целом эти данные свидетельствуют о том, что эффекты метформина на тонкий кишечник могут отличаться от таковых на другие ткани и также могут объяснять профиль побочных эффектов, свойственных этому препарату.
Наиболее распространенным побочным эффектом, ассоциированным с назначением метформина, является диспепсия. Лактатацидоз при лечении метформином регистрируется очень редко (3-9 на 100 тыс. пациенто-лет), и еще реже он вызывает фатальный исход (2-4 на 100 тыс. пациенто-лет). С другой стороны, диспепсические симптомы появляются на раннем этапе терапии, уменьшаются по мере продолжения лечения и полностью разрешаются после его отмены. Эти симптомы, как правило, имеют легкий характер и могут включать диарею, боль в животе, тошноту и рвоту, дисгевзию, вздутие и запор. Точные механизмы их развития не установлены. Предполагается, что они могут возникать вследствие изменений метаболизма серотонина, инкретинов, глюкозы и желчных кислот.
В недавних исследованиях также было продемонстрировано, что метформин изменяет микробный метаболизм в пищеварительном тракте. Это новая интересная гипотеза, которая может объяснять индивидуальные различия в эффектах препарата при диспепсических расстройствах. Karlsson и соавт. изучали состав и функцию фекальной микробиоты у 145 получавших метформин 70-летних европейских женщин с нормальным обменом глюкозы, нарушенной толерантностью к глюкозе и диабетом. Метагеномный анализ пациенток с СД 2 типа показал повышенные уровни представителей семейства Enterobactariaceae (Escherichia, Shigella, Klebsiella и Salmonella) и сниженные уровни Clostridium и Eubacterium. При этом наблюдалась статистически значимая корреляция между cодержанием E. coli и уровнем глюкагоноподобного пептида. Секреция последнего, как известно, снижена у пациенток с СД 2 типа, тогда как метформин повышает его уровни в плазме.
Другими побочными эффектами, ассоциированными с длительной терапией метформином, которые могут объясняться его влиянием на кишечную микробиоту, являются дефицит витамина В12 и фолата, а также изменение метаболизма желчных кислот. Бактерии, в частности Bifidobacterium, являются важными источниками витаминов для макроорганизма. Кроме того, многие анаэробные бактерии участвуют в трансформации желчных кислот в тонком кишечнике, главная функция которых состоит в облегчении метаболизма пищевых жиров и абсорбции жирорастворимых витаминов. В исследовании Napoliatano и соавт. отмена метформина у пациентов с СД 2 типа, находившихся на монотерапии этим препаратом, сопровождалась снижением уровней активного и общего глюкагоноподобного пептида-1 и повышением сывороточных уровней желчных кислот, особенно холевой кислоты и ее конъюгатов. При возобновлении терапии метформином эти эффекты нивелировались. Кроме того, изменения желчных кислот положительно коррелировали с содержанием Firmicutes и отрицательно – с уровнем Bacteroidetes. В целом результаты этого и других современных исследований подтверждают данные ранних работ, в которых внутривенное назначение метформина, несмотря на достижения терапевтически эффективных концентраций в крови, не улучшало контроль глюкозы в отличие от перорального приема препарата.
Выводы
Несмотря на то что метформин на протяжении более полувека широко применяется как эффективный и безопасный препарат для лечения СД 2 типа, открываются все новые и новые механизмы его действия. Антигликемические свойства метформина опосредуются преимущественно подавлением глюконеогенеза в печени, и на сегодня считается, что данный эффект достигается путем ингибирования комплекса I в дыхательной цепи митохондрий. Последующее снижение энергетического статуса клетки непосредственно уменьшает глюконеогенез, снижает действие глюкагона и способствует активации AMPK – главного регулятора клеточного метаболизма; тем не менее пока не установлено, какой из этих трех эффектов вносит наибольший вклад в терапевтическое действие метформина. Кроме того, появляется все больше данных, свидетельствующих о том, что комплекс I является не единственной мишенью метформина; например, недавно было доказано, что метформин ингибирует митохондриальную глицерофосфатдегидрогеназу. Существует высокая вероятность того, что в ближайшем будущем будут идентифицированы новые мишени метформина. Исследование по С. elegans подчеркивает необходимость учитывать действие препаратов не только на макроорганизм, но и на его микробиоту. Бактериальные мишени метформина остаются малоизученными; по-видимому, микробиота посредством неких механизмов может регулировать ряд эффектов препарата на физиологию человека.
В настоящее время метформин одобрен только для лечения СД 2 типа, однако результаты многочисленных исследований, проведенных за последние годы, указывают на огромный терапевтический потенциал этого препарата при других заболеваниях. В настоящем обзоре приводятся различные механизмы, которые могут объяснять эти благоприятные эффекты; в то же время много вопросов остается нерешенными. Очевидно, что лучшее понимание молекулярных путей, задействованных в реализации эффектов метформина, расширит терапевтические горизонты применения этого препарата.
Список литературы находится в редакции.
Pryor R., Cabreiro F. Repurposing metformin:
an old drug with new tricks in its binding pockets.
Biochem J. 2015 Nov 1; 471 (3): 307-22.
Перевел с англ. Алексей Терещенко